Ingredientes ativos: Entecavir
Baraclude 0,5 mg comprimidos revestidos por película
As bulas Baraclude estão disponíveis para os tamanhos de embalagem:- Baraclude 0,5 mg comprimidos revestidos por película
- Baraclude 1 mg comprimidos revestidos por película
- Baraclude 0,05 mg / ml solução oral
Indicações Por que o Baraclude é usado? Para que serve?
Baraclude comprimidos é um medicamento antiviral usado em adultos para tratar a infecção crônica (de longo prazo) pelo vírus da hepatite B. Baraclude pode ser usado em pessoas cujo fígado está danificado, mas ainda está funcionando corretamente (doença hepática compensada) e em pessoas cujo fígado está danificado e não funciona adequadamente (doença hepática descompensada).
Os comprimidos de Baraclude também são usados para tratar a infecção crônica (de longo prazo) pelo vírus da hepatite B em crianças e adolescentes de 2 a 18 anos de idade. Baraclude pode ser utilizado em crianças cujo fígado está danificado, mas ainda funciona bem (doença hepática compensada).
A infecção pelo vírus da hepatite B pode causar danos ao fígado. Baraclude reduz a quantidade de vírus no corpo e melhora a condição do fígado.
Contra-indicações Quando Baraclude não deve ser usado
Não tome Baraclude se tem alergia (hipersensibilidade) ao entecavir ou a qualquer outro componente deste medicamento (listados na secção 6).
Precauções de uso O que você precisa saber antes de tomar Baraclude
Fale com o seu médico ou farmacêutico antes de tomar Baraclude
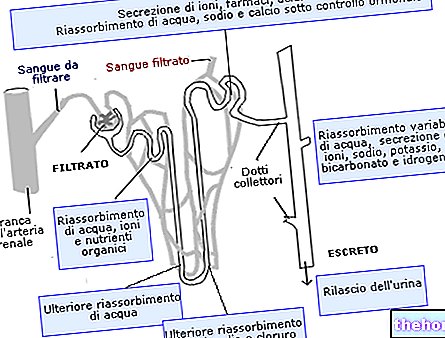
- se já teve problemas renais, informe o seu médico. Isto é importante porque o Baraclude é eliminado do corpo pelos rins e pode necessitar de ajuste posológico ou regime.
- não pare de tomar Baraclude sem o conselho do seu médico, pois a hepatite pode piorar após a interrupção do tratamento. Se o tratamento com Baraclude for interrompido, o seu médico continuará a monitorá-lo e a fazer análises ao sangue durante vários meses.
- fale com o seu médico se o seu fígado está a funcionar bem e, caso contrário, quais os efeitos que podem ter no seu tratamento com Baraclude.
- se também estiver infectado com o vírus VIH (vírus da imunodeficiência humana), informe o seu médico. Não deve tomar Baraclude para tratar a infecção da hepatite B, a menos que já esteja a tomar medicamentos para o VIH, uma vez que a eficácia do futuro tratamento para o VIH pode ser reduzida. Baraclude não verificará se há infecção pelo HIV.
- Tomar Baraclude não o impedirá de infectar outras pessoas com o vírus da hepatite B (VHB) através de relações sexuais ou fluidos corporais (incluindo contaminação com sangue). Por este motivo, é importante tomar precauções para evitar que outras pessoas sejam infectadas com o vírus. de hepatite B (HBV). Uma vacina está disponível para proteger aqueles que estão em risco de contrair a infecção pelo vírus da hepatite B (VHB).
- Baraclude pertence a uma classe de medicamentos que podem causar acidose láctica (excesso de ácido láctico no sangue) e aumento do fígado. Sintomas como náuseas, vômitos e dores de estômago podem indicar o desenvolvimento de acidose láctica. Este efeito colateral raro, mas sério, é ocasionalmente fatal. A acidose láctica é mais comum em mulheres, especialmente se elas estiverem com muito peso. Seu médico irá examiná-la regularmente enquanto você estiver sendo tratado com Baraclude.
- se você já recebeu tratamento para hepatite B crônica, informe o seu médico.
Crianças e adolescentes
Baraclude não deve ser administrado a crianças com menos de 2 anos de idade ou com peso inferior a 10 kg.
Interações Quais medicamentos ou alimentos podem alterar o efeito de Baraclude
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente, ou se vier a tomar outros medicamentos.
Baraclude com comida e bebida
Na maioria dos casos, você pode tomar Baraclude com ou sem alimentos. No entanto, se já foi tratado com um medicamento contendo a substância ativa lamivudina, terá de considerar o seguinte. Se mudou para o tratamento com Baraclude porque o tratamento com lamivudina não teve sucesso, terá de tomar Baraclude uma vez por dia com o estômago vazio. Se a sua doença hepática estiver muito avançada, o seu médico irá receitar-lhe que tome Baraclude com o estômago vazio. Um estômago vazio é definido como pelo menos 2 horas após uma refeição e pelo menos 2 horas antes da próxima refeição.
Crianças e adolescentes (2 a 18 anos de idade) podem tomar Baraclude com ou sem alimentos.
Avisos É importante saber que:
Gravidez, amamentação e fertilidade
Informe o seu médico se estiver grávida ou planejando engravidar. Não foi provado que o uso de Baraclude durante a gravidez seja seguro. A menos que especificamente indicado pelo seu médico, Baraclude não deve ser usado durante a gravidez. É importante que as mulheres com potencial para engravidar utilizem um método eficaz durante o tratamento com Baraclude. para evitar a gravidez.
Você não deve amamentar durante a terapia com Baraclude. Informe o seu médico se estiver amamentando. Não se sabe se o entecavir, a substância ativa do Baraclude, é excretado no leite materno.
Condução e utilização de máquinas
Tonturas, fadiga e sonolência são efeitos secundários frequentes que podem prejudicar a sua capacidade de conduzir e utilizar máquinas. Para qualquer esclarecimento, consulte o seu médico.
Baraclude contém lactose
Este medicamento contém lactose. Se foi informado pelo seu médico que tem “intolerância a alguns açúcares, contacte-o antes de tomar este medicamento.
Dose, Método e Tempo de Administração Como usar Baraclude: Posologia
Nem todos os pacientes precisam tomar a mesma dose de Baraclude.
Tome este medicamento sempre de acordo com as indicações do médico. Em caso de dúvida, consulte o seu médico ou farmacêutico.
Para adultos, a dose recomendada é de 0,5 mg ou 1 mg uma vez ao dia por via oral (por via oral).
Sua dose dependerá de:
- se já recebeu tratamento para a infecção pelo vírus da hepatite B (VHB) e com que medicamento foi tratado.
- se tiver problemas renais. O seu médico pode prescrever uma dose mais baixa ou dizer-lhe para tomá-la menos de uma vez por dia.
- a condição do seu fígado.
Para crianças e adolescentes (2 a 18 anos de idade), o médico do seu filho decidirá a dose certa com base no peso do seu filho.
Baraclude solução oral é recomendado para pacientes com peso entre 10 kg e 32,5 kg.
Crianças com peso mínimo de 32,6 kg podem tomar a solução oral ou comprimidos de 0,5 mg.
Cada dosagem será administrada uma vez ao dia por via oral (boca).
Não há recomendações para Baraclude em crianças menores de 2 anos de idade ou com peso inferior a 10 kg.
O seu médico irá aconselhá-lo sobre a dosagem correta. Para que o medicamento seja totalmente eficaz e reduza o desenvolvimento de resistência à terapia, tome sempre a dose recomendada pelo seu médico. Tome Baraclude enquanto o seu médico lhe disser. O seu médico irá informá-lo se e quando deve interromper o tratamento.
Alguns pacientes devem tomar Baraclude com o estômago vazio (ver Baraclude com alimentos e bebidas na seção 2). Se o seu médico lhe disse para tomar Baraclude com o estômago vazio, isso significa pelo menos 2 horas após uma refeição e pelo menos 2 horas antes da sua próxima refeição.
Se você esquecer de tomar Baraclude
É importante que você não perca nenhuma dose. Se você esquecer de uma dose de Baraclude, tome-a o mais rápido possível e, em seguida, tome a próxima dose na hora certa. Se estiver quase na hora da próxima dose, pule a dose esquecida. Espere e tome a próxima dose na hora marcada.
Não tome uma dose a dobrar para compensar uma dose esquecida.
Não pare de tomar Baraclude sem o conselho do seu médico
Muitas pessoas têm sintomas de hepatite muito graves quando param de tomar Baraclude. Informe o seu médico imediatamente se notar qualquer alteração nos sintomas após interromper o tratamento.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
Overdose O que fazer se você tiver tomado muito Baraclude
Se tomar mais Baraclude do que deveria, contacte o seu médico imediatamente.
Efeitos colaterais Quais são os efeitos colaterais do Baraclude
Como todos os medicamentos, este medicamento pode causar efeitos colaterais, embora nem todas as pessoas os tenham.
Os pacientes tratados com Baraclude relataram os seguintes efeitos colaterais:
- comuns (pelo menos 1 em 100 pacientes): dor de cabeça, insônia (incapacidade de dormir), fadiga (cansaço excessivo), tontura, sonolência (sonolência), vômito, diarreia, náusea, dispepsia (indigestão) e níveis elevados de enzimas hepáticas no sangue.
- pouco frequentes (pelo menos 1 em 1.000 doentes): erupção na pele (erupção na pele), queda de cabelo.
- raros (pelo menos 1 em 10.000 pacientes): reação alérgica grave.
Se tiver quaisquer efeitos secundários, fale com o seu médico ou farmacêutico, incluindo quaisquer efeitos secundários possíveis não mencionados neste folheto.
Relatório de efeitos colaterais
Se tiver quaisquer efeitos secundários, fale com o seu médico ou farmacêutico, incluindo quaisquer efeitos secundários possíveis não mencionados neste folheto. Você também pode relatar os efeitos colaterais diretamente através do sistema nacional de notificação listado no Apêndice V. Ao relatar os efeitos colaterais, você pode ajudar a fornecer mais informações sobre a segurança deste medicamento.
Expiração e retenção
Mantenha este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso no frasco, blister ou cartonagem após EXP. O prazo de validade corresponde ao último dia do mês.
Embalagens blister: Não conservar acima de 30 ° C. Armazene na embalagem original.
Embalagem de frasco: Não conservar acima de 25 ° C. Mantenha o frasco bem fechado.
Não deite quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
Prazo "> Outras informações
O que Baraclude contém
- O ingrediente ativo é o entecavir. Cada comprimido revestido por película contém 0,5 mg de entecavir.
- Os outros excipientes são:
- Núcleo do comprimido: crospovidona, lactose monohidratada, estearato de magnésio, celulose microcristalina e povidona.
- Revestimento do comprimido: hipromelose, macrogol 400, dióxido de titânio (E171) e polissorbato 80 (E433).
Descrição da aparência de Baraclude e conteúdo da embalagem
Os comprimidos revestidos por película (comprimidos) são brancos a esbranquiçados e têm uma forma triangular. Eles são marcados com "BMS" de um lado e "1611" do outro.
Baraclude 0,5 mg comprimidos revestidos por película está disponível em embalagens contendo 30 x 1 ou 90 x 1 comprimido revestido por película (em blisters destacáveis para dose unitária) e em frascos contendo 30 comprimidos revestidos por película.
Nem todos os tamanhos de embalagem podem ser comercializados.
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO -
BARACLUDE 0,5 MG COMPRIMIDOS REVESTIDOS COM PELÍCULA
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA -
Cada comprimido contém 0,5 mg de entecavir (na forma monohidratada).
Excipientes com efeitos conhecidos: cada comprimido contém 120,5 mg de lactose
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA -
Comprimido revestido por película (comprimido).
Comprimido de formato triangular branco a esbranquiçado, marcado com "BMS" de um lado e "1611" do outro.
04.0 INFORMAÇÕES CLÍNICAS -
04.1 Indicações terapêuticas -
Baraclude é indicado para o tratamento da infecção crônica pelo vírus da hepatite B (VHB) (ver seção 5.1) em adultos com:
§ doença hepática compensada e evidência de replicação viral ativa, níveis séricos de alanina aminotransferase (ALT) persistentemente elevados e evidência histológica de inflamação ativa e / ou fibrose.
§ doença hepática descompensada (ver seção 4.4)
Para doença hepática compensada e descompensada, esta indicação é baseada em dados clínicos em pacientes não tratados previamente com infecção de nucleosídeos com vírus da hepatite B HBeAg positivo e HBeAg negativo. Para pacientes com hepatite B refratários à lamivudina ver as seções 4.2, 4.4 e 5.1.
Baraclude também é indicado para o tratamento de infecção crônica pelo vírus da hepatite B (VHB) em pacientes pediátricos virgens de nucleosídeos de 2 a 18 anos de idade com doença hepática compensada que têm evidência de replicação viral ativa e níveis séricos de alanina aminotransferase (ALT) persistentemente elevados ou evidência histológica moderada a grave de inflamação ativa e / ou fibrose. Relativamente à decisão de iniciar o tratamento em doentes pediátricos, ver secções 4.2, 4.4 e 5.1
04.2 Posologia e método de administração -
O tratamento deve ser iniciado por um médico com experiência no tratamento da infecção crônica pelo vírus da hepatite B.
Dosagem
Doença hepática compensada
Pacientes nunca tratados com nucleosídeos: A dose recomendada para adultos é de 0,5 mg uma vez ao dia, com ou sem alimentos.
Pacientes refratário para lamivudina (ou seja, com evidência de viremia durante o tratamento com lamivudina ou com a presença de mutações que conferem resistência à lamivudina [LVDr]) (ver seções 4.4 e 5.1): a dose recomendada em adultos é de 1 mg uma vez ao dia para ser tomada com o estômago vazio (mais de 2 horas antes e mais de 2 horas após uma refeição) (ver secção 5.2). Na presença de mutações LVDr, o uso combinado de entecavir mais um segundo agente antiviral (que não apresenta resistência cruzada com lamivudina ou entecavir) deve ser preferido em vez de monoterapia com entecavir (ver secção 4.4).
Doença hepática descompensada
A dose recomendada para doentes adultos com doença hepática descompensada é de 1 mg uma vez por dia, tomado com o estômago vazio (mais de 2 horas antes e mais de 2 horas após uma refeição) (ver secção 5.2). Para pacientes com hepatite B refratários à lamivudina, ver seções 4.4 e 5.1
Duração da terapia
A duração ideal do tratamento é desconhecida. O tratamento pode ser interrompido:
§ Em pacientes adultos com HBeAg positivo, o tratamento deve ser continuado pelo menos até 12 meses após atingir a soroconversão de HBe (perda de HBeAg e negativização do DNA de HBV com aparecimento de anti-HBe em 2 medições séricas consecutivas repetidas pelo menos 3 - 6 meses depois) ou até seroconversão de HBs ou em caso de perda de eficácia (ver secção 4.4).
§ Em pacientes adultos HBeAg negativos, o tratamento deve ser continuado pelo menos até a soroconversão de HBs ou se houver evidência de perda de eficácia. Em tratamentos prolongados por mais de 2 anos, o ajuste é recomendado para confirmar que a continuação da terapia escolhida permanece adequada para o paciente.
Em pacientes com doença hepática descompensada ou cirrose, a descontinuação do tratamento não é recomendada.
População pediátrica
A decisão de tratar pacientes pediátricos deve ser baseada na consideração cuidadosa das necessidades individuais do paciente e referindo-se às diretrizes atuais de tratamento pediátrico, incluindo o valor das informações de histórico histológico. Com a terapia continuada, os benefícios da supressão virológica de longo prazo devem ser pesados contra o risco do tratamento prolongado, incluindo o surgimento de resistência de vísceras da hepatite B.
Os níveis de alanina aminotransferase (ALT) sérica devem ser persistentemente elevados, por pelo menos 6 meses antes do tratamento, em pacientes pediátricos com doença hepática compensada devido à hepatite B crônica com HBeAg positivo e por pelo menos 12 meses em pacientes com infecção HBeAg negativo. Os doentes pediátricos com peso mínimo de 32,6 kg devem receber uma dose diária de um comprimido de 0,5 mg ou 10 ml (0,5 mg) de solução oral, com ou sem alimentos, utilizado em doentes com peso corporal inferior a 32,6 kg.
Duração da terapia em pacientes pediátricos
A duração ideal do tratamento é desconhecida. De acordo com as diretrizes pediátricas atuais, o tratamento pode ser interrompido:
§ Em pacientes pediátricos HBeAg positivos, o tratamento deve ser continuado por pelo menos 12 meses após o desaparecimento do DNA do HBV e da soroconversão do HBeAg (perda de HBeAg e aparecimento de anti HBe em 2 medições séricas consecutivas repetidas pelo menos 3 a 6 meses depois) ou até seroconversão de HBs ou em caso de perda de eficácia Os níveis séricos de alanina aminotransferase (ALT) e DNA do VHB devem ser monitorizados regularmente após a interrupção do tratamento (ver secção 4.4).
§ Em pacientes pediátricos HBeAg negativos, o tratamento deve ser continuado pelo menos até a soroconversão do HBsAg ou se houver evidência de perda de eficácia.
A farmacocinética em pacientes pediátricos com insuficiência renal ou hepática não foi estudada.
Cidadãos idosos: Não é necessário ajuste de dose com base na idade.A dose deve ser ajustada de acordo com a função renal do paciente (ver recomendações de dosagem em insuficiência renal e seção 5.2)
Gênero e raça: Não são necessários ajustes de gênero ou raça.
Falência renal: A depuração de entecavir diminui com a diminuição da depuração da creatinina (ver secção 5.2). Recomenda-se o ajuste da dose em doentes com hemodiálise da depuração da creatinina ou em diálise peritoneal ambulatorial contínua (CAPD). Ao usar Baraclude solução oral, é recomendada uma redução da dose diária, conforme descrito na tabela. Alternativamente, se a solução oral não estiver disponível, a posologia pode ser ajustada aumentando o intervalo entre as doses, também descrito na tabela. As modificações de dose propostas baseiam-se na extrapolação de dados limitados e a sua segurança e eficácia não foram avaliadas clinicamente, pelo que a resposta virológica deve ser cuidadosamente monitorizada.
* para dosagens
** nos dias de hemodiálise, administrar entecavir após a hemodiálise.
Insuficiência Hepática: Não é necessário ajuste da dose em pacientes com insuficiência hepática.
Método de administração
Baraclude deve ser tomado por via oral.
04.3 Contra-indicações -
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados na seção 6.1.
04.4 Advertências especiais e precauções adequadas de uso -
Falência renal: Recomenda-se o ajuste da dose em doentes com insuficiência renal (ver secção 4.2) .As modificações da dose propostas baseiam-se na extrapolação de dados limitados e a segurança e eficácia relacionadas não foram avaliadas clinicamente. Portanto, a resposta virológica deve ser monitorada cuidadosamente.
Surto de hepatiteAs exacerbações caracterizadas por elevações transitórias da ALT sérica são relativamente comuns na hepatite B crônica. Após o início da terapia antiviral, a ALT sérica pode aumentar em alguns pacientes, bem como diminuir os níveis de DNA do VHB (ver seção 4.8). Entre os pacientes tratados com entecavir, os surtos durante o tratamento tiveram um início mediano de 4 a 5 semanas. Em pacientes com doença hepática compensada, essas elevações na ALT sérica geralmente não são acompanhadas por concentrações aumentadas de bilirrubina sérica ou descompensação hepática. Pacientes com doença hepática avançada ou cirrose podem apresentar maior risco de descompensação hepática após uma exacerbação da hepatite e, portanto, precisarão ser monitorados de perto durante a terapia.
Também foi relatada exacerbação aguda de hepatite em pacientes que descontinuaram a terapia para hepatite B (ver seção 4.2). As exacerbações pós-tratamento geralmente estão associadas a níveis elevados de DNA do VHB, e a maioria delas, no entanto, foram observadas exacerbações graves, incluindo mortes .
Entre os doentes tratados com entecavir que nunca receberam nucleósidos, as exacerbações pós-tratamento tiveram uma mediana de início de 23 - 24 semanas e a maioria ocorreu em doentes HBeAG negativos (ver secção 4.8). A função hepática deve ser monitorada em intervalos repetidos com testes clínicos e laboratoriais pelo menos a cada 6 meses após a descontinuação da terapia para hepatite B. Se apropriado, a terapia para hepatite B pode ser reiniciada.
Pacientes com doença hepática descompensada: Uma "alta taxa de eventos adversos hepáticos graves (independentemente da causa) foi observada em pacientes com doença hepática descompensada, particularmente aqueles com doença de Child-Turcotte-Pugh (CTP) classe C, em comparação com as porcentagens encontradas em pacientes com função hepática compensada Além disso, os pacientes com doença hepática descompensada podem ter um risco maior de acidose láctica e eventos adversos renais específicos, como síndrome hepatorrenal. Portanto, os parâmetros clínicos e laboratoriais devem ser monitorados de perto nesta população de pacientes. (Ver também seções 4.8 e 5.1) .
Acidose láctica e hepatomegalia grave com esteatose: acidose láctica (na ausência de hipoxemia), às vezes fatal, geralmente associada a hepatomegalia grave e esteatose hepática, foi relatada com o uso de análogos de nucleosídeos. Como o entecavir é um análogo de nucleosídeo, este risco não pode ser excluído. Tratamento com análogos de nucleosídeo deve ser descontinuado no caso de níveis elevados de aminotransferase, hepatomegalia progressiva ou acidose metabólica / láctica de etiologia desconhecida. Sintomas digestivos benignos, como náuseas, vômitos e dor abdominal, podem indicar o desenvolvimento de acidose láctica. Casos graves, às vezes com desfecho fatal. , foram associados a pancreatite, insuficiência hepática / doença hepática gordurosa, insuficiência renal e níveis elevados de ácido láctico sérico. Deve-se ter cuidado ao administrar análogos de nucleosídeos a pacientes (especialmente mulheres obesas) com hepatomegalia, hepatite ou outros fatores de risco conhecidos para doença hepática Esses pacientes devem ser acompanhados de perto mente.
Para diferenciar as elevações nas aminotransferases devido à resposta ao tratamento daquelas potencialmente relacionadas à acidose láctica, os médicos devem garantir que as alterações na ALT estejam associadas a aumentos em outros marcadores laboratoriais de hepatite B crônica.
Resistência e precaução especial para pacientes refratário para lamivudina: Mutações na polimerase de HBV que decodificam substituições de resistência à lamivudina podem levar à ocorrência subsequente de substituições secundárias, incluindo aquelas associadas à resistência ao entecavir (ETVr). Em uma pequena porcentagem de pacientes refratários à lamivudina, mutações de ETVr para rtT184, rtS202 ou rtM & SUP2; 50, estavam presentes no início do estudo. Pacientes com VHB resistente à lamivudina têm maior risco de desenvolver resistência subsequente ao entecavir em comparação com pacientes não resistentes à lamivudina. A probabilidade cumulativa de emergência de genótipos resistentes ao entecavir após 1, 2, 3, 4 e 5 anos de tratamento em estudos de pacientes refratários à lamivudina foi de 6%, 15%, 36%, 47% e 51%, respectivamente. A resposta virológica deve ser monitorada com frequência na população refratária. A lamivudina e o teste de resistência apropriado devem ser realizados em pacientes com uma resposta virológica subótima após 24 séries Durante o tratamento com entecavir, deve ser considerado o ajuste do tratamento (ver secções 4.5 e 5.1). Ao iniciar a terapia em pacientes com história documentada de VHB resistente à lamivudina, o uso combinado de entecavir mais um segundo agente antiviral (que não apresenta resistência cruzada com lamivudina ou entecavir) deve ser preferido em vez de monoterapia com entecavir. associado a um risco aumentado de resistência subsequente ao entecavir, independentemente do grau de doença hepática; a descoberta virológica pode estar associada a complicações clínicas graves da doença hepática subjacente em pacientes com doença hepática descompensada. com doença hepática descompensada e HBV resistente à lamivudina, o o uso combinado de entecavir mais um segundo agente antiviral (que não apresenta resistência cruzada com lamivudina ou entecavir) deve ser preferível à monoterapia com entecavir.
População pediátrica: Uma taxa de resposta virológica mais baixa (DNA de HBV
Transplante de fígado: A função renal deve ser avaliada cuidadosamente antes e durante a terapia com entecavir em receptores de transplante de fígado recebendo ciclosporina ou tacrolimus (ver seção 5.2).
Coinfecção com hepatite C ou D: Não existem dados sobre a eficácia do entecavir em doentes coinfetados com os vírus da hepatite C ou D.
Pacientes co-infectados com o vírus da imunodeficiência humana (HIV) / VHB que não estão recebendo terapia antirretroviral concomitante: entecavir não foi avaliado em doentes coinfetados por VIH / VHB que não receberam tratamento concomitante eficaz para VIH. Foi observado início de resistência ao VIH quando o entecavir foi utilizado para tratar a infecção por hepatite B crónica. em doentes infetados por VIH que não receberam terapêutica antirretroviral altamente activa ( HAART) (consulte a seção 5.1). Portanto, a terapia com entecavir não deve ser usada em pacientes coinfetados por HIV / VHB que não estejam sendo tratados com HAART. Entecavir não foi estudado para o tratamento da infecção por HIV e não é recomendado para este uso.
Pacientes co-infectados por HIV / VHB recebendo terapia antirretroviral concomitante: o entecavir foi estudado em 68 adultos coinfetados por VIH / VHB a receberem HAART contendo lamivudina (ver secção 5.1). Não há dados disponíveis sobre a eficácia do entecavir em pacientes HBeAg negativos infectados pelo HIV. Existem dados limitados sobre pacientes coinfetados por HIV com contagens baixas de células CD4 (células / mm³).
Em geral: Os pacientes devem ser informados de que a terapia com entecavir não demonstrou reduzir o risco de transmissão do VHB e, portanto, devem ser tomadas as precauções adequadas.
Lactose: Cada dose diária de 0,5 mg deste medicamento contém 120,5 mg de lactose.
Os doentes com problemas hereditários raros de intolerância à galactose, deficiência de lactase de Lapp ou má absorção de glucose-galactose não devem tomar este medicamento. Baraclude solução oral que não contém lactose está disponível para esses indivíduos.
04.5 Interações com outros medicamentos e outras formas de interação -
Uma vez que o entecavir é eliminado principalmente por via renal (ver secção 5.2), a coadministração com medicamentos que reduzem a função renal ou competem com a secreção tubular ativa pode aumentar as concentrações séricas de ambos os medicamentos. Para além da lamivudina, adefovir dipivoxil e tenofovir disoproxil fumarato, os efeitos da coadministração de entecavir com medicamentos que são eliminados por via renal ou que afetam a função renal não foram avaliados. Os doentes devem ser cuidadosamente monitorizados quanto a efeitos indesejáveis que possam surgir durante a coadministração de entecavir com esses medicamentos.
Não foram observadas interações farmacocinéticas entre entecavir e lamivudina, adefovir ou tenofovir.
O entecavir não é um substrato, indutor ou inibidor das enzimas do citocromo P450 (CYP450) (ver secção 5.2). Portanto, é improvável que ocorram interações medicamentosas transportadas pelo CYP450 com o entecavir.
População pediátrica
Os estudos de interação foram realizados apenas em adultos.
04.6 Gravidez e amamentação -
Mulheres com potencial para engravidar: Como os riscos potenciais para o desenvolvimento fetal são desconhecidos, as mulheres com potencial para engravidar devem usar métodos contracetivos eficazes.
Gravidez: Não existem estudos adequados sobre a utilização de entecavir em mulheres grávidas Os estudos em animais revelaram toxicidade reprodutiva em doses elevadas (ver secção 5.3).
O risco potencial para o ser humano é desconhecido. Baraclude não deve ser usado durante a gravidez, a menos que seja claramente necessário. Não existem dados sobre os efeitos do entecavir na transmissão do VHB da mãe para o recém-nascido, pelo que devem ser tomadas medidas adequadas para prevenir a aquisição neonatal do VHB.
Hora da alimentação: não se sabe se o entecavir é excretado no leite humano. Os dados toxicológicos disponíveis em animais mostraram excreção de entecavir no leite materno (para detalhes ver secção 5.3). Um risco para as crianças não pode ser excluído. A amamentação deve ser interrompida durante a terapia com Baraclude.
Fertilidade: Os estudos de toxicologia em animais administrados com entecavir não mostraram evidência de perda de fertilidade (ver secção 5.3).
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas -
Não foram realizados estudos sobre a capacidade de conduzir e utilizar máquinas.Tonturas, fadiga e sonolência são efeitos secundários frequentes que podem prejudicar a capacidade de conduzir e utilizar máquinas.
04.8 Efeitos indesejáveis -
para. Resumo do perfil de segurança
Em estudos clínicos de pacientes com doença hepática compensada, as reações adversas mais comuns de qualquer gravidade, com pelo menos uma relação possível com o entecavir, foram: cefaleia (9%), fadiga (6%), tonturas (4%) e náuseas ( 3%). Foi notificada exacerbação de hepatite durante e após a descontinuação da terapêutica com entecavir (ver secção 4.4 c. Descrição das reações adversas selecionadas).
b. Lista de reações adversas
A avaliação das reações adversas é baseada na experiência de vigilância pós-comercialização e em quatro estudos clínicos nos quais 1.720 pacientes com infecção crônica pelo vírus da hepatite B e doença hepática compensada foram tratados de forma duplo-cega com entecavir (n = 862) ou lamivudina. (N = 858) até 107 semanas (ver seção 5.1). Nestes estudos, os perfis de segurança, incluindo alterações nos parâmetros laboratoriais, foram semelhantes para entecavir 0,5 mg uma vez ao dia (679 doentes HBeAg virgens de nucleósidos positivos ou negativos durante uma média de 53 semanas), entecavir 1 mg uma vez por dia (183 refractários à lamivudina doentes tratados durante uma média de 69 semanas) e lamivudina.
As reações adversas consideradas pelo menos possivelmente relacionadas com o tratamento com entecavir são listadas por classes de sistemas de órgãos. A frequência é definida como muito comum (≥ 1/10); comum (≥ 1/100 a
Distúrbios do sistema imunológico: raro: reação anafilactoide
Distúrbios psiquiátricos: comum: insônia
Doenças do sistema nervoso: comum: dor de cabeça, tontura, sonolência
Problemas gastrointestinais: comuns: vômitos, diarreia, náuseas, dispepsia
Doenças hepatobiliares comum: elevação das transaminases
Doenças do tecido cutâneo e subcutâneo: incomum: erupção cutânea, alopecia
Perturbações gerais e condições no local de administração: comum: fadiga
Foram notificados casos de acidose láctica, frequentemente associada a descompensação hepática, outras condições médicas graves ou exposição ao medicamento (ver secção 4.4).
Tratamento além de 48 semanas: o tratamento continuado com entecavir por uma duração média de 96 semanas não mostrou novos sinais de segurança.
c. Descrição das reações adversas selecionadas
Anormalidades nos testes laboratoriais: em ensaios clínicos em pacientes virgens de nucleosídeos, 5% tiveram elevações de ALT> 3 vezes da linha de base e 2 vezes da linha de base dos pacientes, juntamente com bilirrubina total> limites de 2 vezes acima do normal (Limite superior do normal, ULN ) e> 2 vezes os valores da linha de base. Níveis de albumina amilase> 3 vezes a linha de base em 2%, níveis de lipase> 3 vezes a linha de base em 11% e plaquetas
Em ensaios clínicos em doentes refratários à lamivudina, 4% tiveram elevações de ALT> 3 vezes da linha de base e uma percentagem de 2 vezes da linha de base, juntamente com bilirrubina total> 2 vezes o LSN e> 2 vezes em comparação com os valores da linha de base. Níveis de amilase> 3 vezes o valor basal ocorreram em 2% dos pacientes, os níveis de lipase> 3 vezes o valor basal em 18% e plaquetas
Surtos durante o tratamento: em estudos de pacientes virgens de nucleosídeos, aumentos de ALT> 10 vezes o LSN e> 2 vezes o valor basal ocorreram durante o tratamento em 2% dos pacientes tratados com entecavir versus 4% dos pacientes tratados com lamivudina. Em estudos de pacientes refratários à lamivudina, elevações de ALT> 10 vezes o LSN e> 2 vezes o valor basal ocorreram durante o tratamento em 2% dos pacientes tratados com entecavir versus 11% dos pacientes tratados com lamivudina. Entre os pacientes tratados com entecavir, elevações de ALT durante o tratamento tiveram um tempo médio para elevação de 4 a 5 semanas, geralmente resolvido com a continuação do tratamento e, na maioria dos casos, foram associados com diminuição da carga viral ≥ 2 log10 / ml que precedeu ou coincidiu com a elevação de ALT. Durante o tratamento, recomenda-se a monitorização periódica da função hepática.
Exacerbações após a descontinuação do tratamento: foram relatadas exacerbações agudas de hepatite em pacientes que descontinuaram o tratamento para o vírus da hepatite B, incluindo terapia com entecavir (ver seção 4.4). Nucleosídeos, 6% dos pacientes tratados com entecavir e 10% dos pacientes tratados com lamivudina experimentaram Elevações de ALT (> 10 vezes o LSN e> 2 vezes os valores de referência [valores mínimos na linha de base ou medições na "última dose administrada]) durante o acompanhamento pós-tratamento. Entre os pacientes virgens de nucleosídeo tratados com entecavir, as elevações de ALT tiveram um tempo médio de elevação de 23-24 semanas, e 86% (24/28) das elevações de ALT ocorreram em pacientes HBeAg negativos. Estudos em pacientes refratários à lamivudina, apenas um número limitado de pacientes teve acompanhamento, 11% dos pacientes tratados com entecavir e nenhum dos pacientes tratados com lamivudina desenvolveram elevações de ALT durante o acompanhamento pós-tratamento.
Em ensaios clínicos, o tratamento com entecavir era interrompido se os doentes obtivessem uma resposta pré-específica. Se o tratamento for interrompido independentemente da resposta à terapia, a taxa de elevações de ALT após o tratamento pode ser maior.
d. População pediátrica
A segurança de entecavir em pacientes pediátricos de 2 a 8 anos de idade é baseada em dois ensaios clínicos em andamento em indivíduos cronicamente infectados pelo VHB; um estudo farmacocinético de Fase 2 (estudo 028) e um estudo de Fase 3 (estudo 189). Esses estudos envolveram 173 indivíduos HBeAg positivos que nunca haviam sido tratados com nucleosídeos antes e foram tratados com entecavir por um período médio de 60 semanas. As reações adversas observadas em indivíduos pediátricos tratados com entecavir foram consistentes com as observadas em ensaios clínicos de entecavir em adultos (ver um Resumo do perfil de segurança e secção 5.1).
E. Outras populações especiais
Experiência em pacientes com doença hepática descompensada: O perfil de segurança do entecavir em pacientes com doença hepática descompensada foi avaliado em um estudo comparativo aberto e randomizado no qual os pacientes foram tratados com entecavir 1 mg / dia (n = 102) ou adefovir dipivoxil 10 mg / dia (n = 89) (estudo 048). Em comparação com as reações adversas relatadas na seção b. Lista de reações adversas, uma "reação adversa adicional [diminuição do bicarbonato no sangue (2%)] foi observada em pacientes tratados com entecavir até a semana 48. A taxa de mortalidade cumulativa ao longo do estudo foi de 23% (23/102) e as causas de as mortes foram geralmente relacionadas ao fígado, como esperado nesta população. A taxa cumulativa de carcinoma hepatocelular (CHC) ao longo do estudo foi de 12% (12/102). Os eventos adversos graves foram geralmente relacionados ao fígado, com uma frequência cumulativa de 69% ao longo do estudo Os doentes com uma pontuação CTP elevada no início do estudo tinham um risco aumentado de desenvolver acontecimentos adversos graves (ver secção 4.4).
Alterações nos testes laboratoriais: entre os pacientes com doença hepática descompensada tratados com entecavir até a semana 48, nenhum apresentou elevações de ALT> 10 vezes o limite superior do normal (LSN) e> 2 vezes o valor basal, el "1% dos pacientes teve elevações de ALT> 2 vezes linha de base junto com bilirrubina total> 2 vezes o limite superior do normal (LSN) e> 2 vezes a linha de base. Níveis de albumina 3 vezes a linha de base. valores basais em 10% e plaquetas
Experiência em pacientes coinfetados por HIV: O perfil de segurança de entecavir em um número limitado de pacientes coinfetados por HIV / VHB em tratamento HAART (terapia antirretroviral altamente ativa) foi semelhante ao perfil de segurança de pacientes monoinfetados com VHB (ver seção 4.4 )
Sexo / idade: Não houve diferença aparente no perfil de segurança do entecavir no que diz respeito ao sexo (≈ 25% mulheres em ensaios clínicos) ou idade (≈ 5% dos doentes> 65 anos de idade).
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas ocorridas após a autorização do medicamento é importante porque permite a monitorização contínua da relação benefício / risco do medicamento.Os profissionais de saúde são convidados a notificar quaisquer suspeitas de reações adversas através do sistema nacional de notificação.
04.9 Overdose -
Existem relatos limitados de sobredosagem com entecavir em doentes. Indivíduos saudáveis que receberam até 20 mg / dia por 14 dias e doses únicas de até 40 mg não tiveram reações adversas inesperadas. Em caso de sobredosagem, o paciente deve ser monitorado quanto a sinais de toxicidade e receber tratamento de suporte padrão apropriado.
05.0 PROPRIEDADES FARMACOLÓGICAS -
05.1 "Propriedades farmacodinâmicas -
Grupo farmacoterapêutico: antivirais para uso sistêmico, nucleosídeos e nucleotídeos inibidores da transcriptase reversa
Código ATC: J05AF10
Mecanismo de ação: o entecavir, um análogo do nucleosídeo da guanosina ativo contra a polimerase do VHB, é fosforilado com eficiência na forma trifosfato (TP) ativa, que tem meia-vida intracelular de 15 horas. Competindo com o substrato natural deoxiguanosina TP, entecavir-TP funcionalmente, inibe as 3 atividades da polimerase viral: iniciação da polimerase do HBV, transcrição reversa da fita negativa do DNA a partir do RNA mensageiro pré-demônico e síntese da fita positiva do DNA do HBV. O Ki do entecavir-TP para a DNA polimerase do HBV é 0,0012 mcM. O entecavir-TP é um inibidor fraco da DNA polimerase celular α, β e δ com valores de Ki de 18 a 40 mcM. Além disso, altas exposições ao entecavir não são significativas efeitos adversos em γ polimerases ou síntese de DNA mitocondrial em células HepG2 (Ki> 160 mcM).
Atividade antiviral: o entecavir inibiu a síntese de DNA do VHB (redução de 50%, EC50) a uma concentração de 0,004 μM em células HepG2 humanas transinfetadas com HBV de tipo selvagem. O valor médio de EC50 para entecavir versus LVDr HBV (rtL 180M e rtM204V) foi de 0,026 μM (intervalo de 0,010 - 0,059 μM) Os vírus recombinantes com substituições resistentes ao adefovir rtN236T ou rtA181V permaneceram totalmente suscetíveis ao entecavir.
Uma análise da atividade inibitória do entecavir contra um painel de laboratório e isolados clínicos de HIV-1 realizada usando diferentes células e métodos produziram valores de EC50 variando de 0,026 a> 10 μM; os menores valores de EC50 foram observados quando no teste Níveis baixos de Em cultura de células, o entecavir selecionou uma substituição M184I em concentrações micromolares, confirmando a pressão inibitória do entecavir em concentrações elevadas. As variantes do VIH contendo a substituição M184V mostraram perda de susceptibilidade ao entecavir (ver secção 4.4).
No ensaio de combinação de HBV em cultura de células, abacavir, didanosina, lamivudina, estavudina, tenofovir ou zidovudina não foram antagonistas da atividade anti-VHB do entecavir em uma grande porcentagem das concentrações. No ensaio antiviral para HIV, o entecavir em concentrações micromolares não foi antagonista para a atividade anti-HIV em cultura de células desses seis NRTIs ou emtricitabina.
Resistência em cultura de células: em relação ao HBV de tipo selvagem, os vírus LVDr contendo as substituições rtM204V e rtL180M na transcriptase reversa mostram uma diminuição de 8 vezes na suscetibilidade ao entecavir. A incorporação de outras substituições de aminoácidos rtT184, rtS202 e / ou rtM250 causa diminuição da suscetibilidade ao entecavir em cultura de células. As substituições observadas em isolados clínicos (rtT184A, C, F, G, I, L, M ou S; rtS202 C, G ou I; e / ou rtM250I, L ou V) resultaram em uma diminuição adicional de 16 a 741 vezes na suscetibilidade ao entecavir em comparação com o vírus do tipo selvagem. As substituições únicas de ETVr (resistência ao entecavir) rtT184, rtS202 e rtM250 têm apenas um efeito modesto na suscetibilidade ao entecavir e não foram observadas na ausência de substituições LVDr (resistência à lamivudina) em mais de 1000 amostras de pacientes. A resistência é mediada pela redução da ligação inibitória ao inv prejudicado ersa de HBV e HBV resistente exibem capacidade de replicação reduzida em cultura de células.
Experiência clínica: A demonstração do benefício é baseada nas respostas histológicas, virológicas, bioquímicas e serológicas após 48 semanas de tratamento em ensaios clínicos controlados ativos de 1.633 adultos com infecção crônica de hepatite B, evidência de replicação viral e doença hepática compensada. A segurança e eficácia do entecavir também foram avaliadas em um estudo clínico controlado em 191 pacientes infectados por VHB com doença hepática descompensada e em um estudo clínico em 68 pacientes coinfetados por VHB e HIV.
Em estudos em pacientes com doença hepática compensada, melhora histológica definida como redução ≥ 2 pontos no índice necroinflamatório de Knodell desde o início sem piora no escore de fibrose de Knodell. As respostas para pacientes com escore de fibrose de Knodell de 4 no início (cirrose) foram comparáveis com todas as respostas em todas as medidas de eficácia (todos os pacientes tinham doença hepática compensada). As pontuações do índice de atividade histológica (HAI) de Knodell de base elevada (> 10) foram associadas a uma maior melhora histológica em pacientes virgens de nucleosídeo. Níveis basais de ALT ≥ 2 vezes a linha de base em pacientes HBeAg positivos virgens de nucleosídeo limite superior de DNA de HBV normal e basal ≤ 9,0 log 10 cópias / mL foram ambos associados a altas taxas de resposta virológica (semana 48 HBV DNA
Experiência em pacientes virgens de nucleosídeo com doença hepática compensada: Os resultados de ensaios clínicos duplo-cegos randomizados de 48 semanas comparando entecavir (ETV) e lamivudina (LVD) em pacientes HBeAg positivos e HBeAg negativos são mostrados na tabela abaixo:
* valor p vs lamivudina
pacientes com histologia avaliável no início do estudo (pontuação necroinflamatória de Knodell basal ≥ 2)
b objetivo principal
Ensaio PCR cRoche Cobas Amplicor (LLOQ = 300 cópias / mL)
Experiência em pacientes refratário à lamivudina com doença hepática compensada: Em um estudo duplo-cego randomizado de pacientes HBeAg positivos refratários à lamivudina, com 85% dos pacientes com mutações LVDr no início do estudo, os pacientes que tomavam lamivudina no início do estudo foram trocados para entecavir 1 mg uma vez ao dia, sem período de washout ou sobreposição ( n = 141), ou continuado com lamivudina 100 mg uma vez ao dia (n = 145). Os resultados em 48 semanas são mostrados na tabela abaixo.
* valor p vs lamivudina
pacientes com histologia avaliável no início do estudo (pontuação necroinflamatória de Knodell basal ≥ 2)
b objetivo principal
Ensaio PCR cRoche Cobas Amplicor (LLOQ = 300 cópias / mL)
Resultados além de 48 semanas de tratamento:
O tratamento foi interrompido quando os critérios de resposta pré-específicos foram atendidos em 48 semanas ou durante o segundo ano de terapia. Os critérios de resposta foram a supressão virológica de HBV (HBV DNA
Pacientes nunca tratados com nucleosídeos:
HBeAg positivo (estudo 022): o tratamento com entecavir por até 96 semanas (n = 354) resultou em uma taxa de resposta cumulativa de 80% para o DNA do VHB
No final da dosagem, entre os pacientes que continuaram o tratamento além de 52 semanas (mediana de 96 semanas), 81% dos 243 pacientes tratados com entecavir e 39% dos 164 pacientes tratados com lamivudina tinham DNA do VHB.
AgHBe negativo (estudo 027): O tratamento com entecavir até 96 semanas (n = 325) resultou em uma taxa de resposta cumulativa de 94% para o DNA do VHB
Para os 26 pacientes tratados com entecavir e 28 pacientes tratados com lamivudina que continuaram o tratamento além de 52 semanas (mediana de 96 semanas), 96% dos pacientes tratados com entecavir e 64% dos pacientes tratados com lamivudina no final da dosagem tinham DNA do VHB
Para os pacientes que responderam ao protocolo, o critério de resposta foi mantido por até 24 semanas de acompanhamento após o tratamento em 75% (83/111) daqueles que responderam ao tratamento com entecavir em 73.% (68/93) daqueles que responderam ao tratamento com lamivudina no estudo 022 e 46% (131/286) daqueles que responderam ao entecavir versus 31% (79/253) daqueles que responderam ao tratamento com lamivudina no estudo 027. Dentro de 48 semanas de acompanhamento após o Ao final do tratamento, um número substancial de pacientes HBeAg negativos perderam a resposta.
Resultados da biópsia hepática: 57 pacientes de estudos essenciais conduzidos em pacientes virgens de nucleosídeo 022 (HBeAg positivo) e 027 (HBeAg negativo), inscritos em um estudo de rolar a longo prazo, foram avaliados nos resultados histológicos hepáticos a longo prazo. Nos estudos principais, a dose de entecavir foi de 0,5 mg por dia (exposição média de 85 semanas) e no estudo principal rolar de 1 mg por dia (exposição média de 177 semanas) e inicialmente 51 pacientes no rolar eles também receberam lamivudina (duração média de 29 semanas). Destes pacientes, 55/57 (96%) tiveram melhora histológica conforme definido acima (ver acima) e 50/57 (88%) tiveram diminuição da pontuação de fibrose de Ishak ≥ 1 ponto. Entre os pacientes com pontuação de fibrose de Ishak ≥ 2 no início do estudo, 25/43 (58%) mostraram uma diminuição de ≥ 2 pontos. Todos os pacientes (10/10) com fibrose basal ou cirrose avançada (escore de fibrose Ishak de 4, 5 ou 6) tiveram uma diminuição ≥ 1 ponto (diminuição média desde o início foi de 1,5 pontos). No momento da biópsia de longo prazo, todos os pacientes apresentavam DNA de HBV
Refratários para lamivudina:
HBeAg positivo (estudo 026): O tratamento com entecavir por até 96 semanas (n = 141) resultou em uma taxa de resposta cumulativa de 30% para a conversão de HBeAg sérico de DNA de HBV.
Para os 77 pacientes que continuaram o tratamento com entecavir além de 52 semanas (mediana de 96 semanas), 40% dos pacientes apresentaram DNA do VHB
Idade gênero:
Não houve diferença aparente na eficácia do entecavir no que diz respeito às diferenças de gênero (≈ 25% mulheres em ensaios clínicos) ou idade (≈ 5% dos pacientes> 65 anos).
Populações especiais
Pacientes com doença hepática descompensada: No estudo 048, 191 pacientes com infecção VHB crônica positiva ou negativa para HBeAg e evidência de descompensação hepática, definida como uma pontuação de CTP de 7 ou maior, receberam entecavir 1 mg uma vez ao dia ou adefovir dipivoxil 10 mg uma vez ao dia. Os doentes nunca receberam tratamento para o VHB ou já foram pré-tratados (excluindo pré-tratamento com entecavir, adefovir dipivoxil ou tenofovir disoproxil fumarato). No início do estudo, os pacientes tinham uma pontuação CTP média de 8,59 e 26% dos pacientes eram da classe C de CTP. A pontuação média do Modelo para Doença Hepática em Estágio Final (MELD) no início do estudo era 16,23. A concentração média de DNA de VHB no soro por PCR foi de 7,83 log10 cópias / mL e a concentração média de ALT no soro foi de 100 U / L; 54% dos pacientes eram HBeAg positivos e 35% dos pacientes tinham substituições LVDr em Entecavir foi superior a adefovir dipivoxil em o endpoint primário de eficácia avaliando as mudanças médias desde a linha de base na concentração sérica de HBV DNA por PCR na semana 24. Os resultados dos endpoints do estudo selecionado nas semanas 24 e 48 são mostrados na tabela.
um Ensaio de PCR Roche COBAS Amplicor (LLOQ = 300 cópias / mL).
bNC = F (paciente que não completou = falha), significa que o tratamento foi interrompido antes da semana de análise, incluindo motivos como morte, falta de eficácia, eventos adversos, falta de adesão / perda de acompanhamento, considerados como falhas (por exemplo, DNA de HBV ≥ 300 cópias / mL)
cNC = M (pacientes não concluídos = ausentes)
d Definido como uma redução ou nenhuma mudança da linha de base na pontuação CTP
A pontuação do eMean MELD no início do estudo foi de 17,1 para ETV e 15,3 para adefovir dipivoxil.
f O denominador é o número de pacientes com valores anormais na linha de base.
* p
ULN = limite superior da norma, LLN = limite inferior da norma.
O tempo para o início do CHC ou morte (o que ocorrer primeiro) foi comparável nos dois grupos de tratamento; as taxas de mortalidade cumulativas ao longo do estudo foram 23% (23/102) e 33% (29/89) para pacientes tratados com entecavir e adefovir dipivoxil, respectivamente, e as taxas cumulativas de HCC ao longo do estudo foram de 12% ( 12/102) e 20% (18/89) para entecavir e adefovir dipivoxil, respectivamente.
Para pacientes com substituições LVDr basais, a porcentagem de pacientes com DNA de HBV
Pacientes co-infectados por HIV / VHB tratados concomitantemente com HAART: O estudo 038 incluiu 67 pacientes HBeAg positivos e 1 HBeAg negativos co-infectados com HIV. Os pacientes exibiram HIV estável e controlado (HIV RNA placebo (n = 17) por 24 semanas, seguido por mais 24 semanas em que todos os pacientes receberam entecavir. Em 24 semanas, a redução da carga viral do VHB foi significativamente maior. Com entecavir (-3,65 vs um aumento de 0,11 log10 cópias / ml). Para pacientes originalmente designados para tratamento com entecavir, a redução no DNA do VHB em 48 semanas foi de -4,20 log10 cópias / ml, a normalização de ALT apareceu em 37% dos pacientes com anormalidades basais de ALT e nenhuma alcançou a soroconversão de HBeAg.
Pacientes coinfetados por HIV / VHB não concomitantes com terapia HAART: o entecavir não foi avaliado em doentes coinfetados por VIH / VHB que não receberam tratamento concomitante eficaz para o VIH. Foram notificadas reduções no ARN do VIH em doentes coinfetados por VIH / VHB a receber entecavir em monoterapia sem HAART. Em alguns casos, foi observada a seleção da variante do HIV M184V, o que pode ter implicações na seleção de regimes HAART que o paciente pode tomar no futuro. Portanto, o entecavir não deve ser usado neste tipo de população devido ao potencial desenvolvimento de resistência ao VIH (ver secção 4.4).
Transplante de fígado: A segurança e eficácia de entecavir 1 mg uma vez ao dia foram avaliadas em um estudo de braço único em 65 pacientes que receberam transplante de fígado para complicações de infecção VHB crônica e apresentaram DNA de HBV branco e 37% asiático, com média de idade de 49 anos: 89% dos pacientes eram HBeAg-negativos no momento do transplante. Dos 61 pacientes avaliados quanto à eficácia (eles receberam entecavir por pelo menos 1 mês), 60 também receberam imunoglobulina contra hepatite B (HBIg) como parte do regime de profilaxia pós-transplante. desses 60 pacientes, 49 receberam terapia com HBIg por mais de 6 meses. Na semana 72 após o transplante, nenhum dos 55 casos observados mostrou reativação da replicação viral [definida como DNA de HBV ≥ 50 UI / ml (aproximadamente 300 cópias / ml)] , e nenhuma reativação virológica de HBV foi relatada nos 6 pacientes excluídos restantes. Todos os 61 pacientes apresentaram perda de HBsAg pós-transplante, e 2 desses tornaram-se HBsAg positivos, mantendo níveis indetectáveis de DNA de HBV (
População pediátrica: O Estudo 189 é um estudo em andamento com base na eficácia e segurança do entecavir, envolvendo 180 crianças e adolescentes, de 2 a 18 anos de idade, não tratados previamente com nucleosídeos e sofrendo de hepatite B crônica com HBeAg positivo, doença hepática compensada e ALT elevada Os indivíduos foram randomizados (2: 1) para receber tratamento cego com entecavir de 0,015 mg / kg a 0,5 mg / dia (N = 120) ou placebo. (N = 60). A randomização foi estratificada por faixa etária (2 a 6 anos ;> 6 a 12 anos; e> 12 a
24% (20/82) dos indivíduos no grupo de entecavir e 2% (1/41) dos indivíduos no grupo de placebo alcançaram o desfecho principal. Em 48 semanas, 46% (38/82) dos indivíduos recebendo entecavir e 2% (1/41) dos indivíduos que receberam placebo alcançaram valores de DNA de HBV
Nos 2 estudos pediátricos (estudos 028 e 189), 110 pacientes que receberam entecavir por até 48 semanas foram monitorados quanto à resistência. Avaliações genotípicas foram realizadas em todos os pacientes que tiveram avanço virológico ou DNA do VHB ≥ 50 UI / mL em 48 semanas ou que interromperam a terapia precocemente. Nenhuma substituição de aminoácido associada à resistência ao entecavir foi identificada.
Resistência clínica: os doentes inicialmente tratados em ensaios clínicos com entecavir 0,5 mg (nunca antes de nucleósidos) ou 1,0 mg (refractário à lamivudina) e com uma medição do ADN do VHB de 24 semanas por PCR durante ou depois da terapêutica, foram monitorizados quanto à resistência. estudos até 240 semanas, em pacientes virgens de nucleosídeo, evidência genotípica de substituições de ETVr em rtT184, rtS202 ou rtM250 foi identificada em 3 dos pacientes que receberam entecavir., 2 dos quais também tiveram avanço virológico (ver tabela). Essas substituições foram observadas apenas na presença de substituições LVDr (rtM204V e rtL180M).
a Os resultados refletem o uso de uma dose de 1 mg de entecavir para 147 de 149 pacientes no ano 3, para todos os pacientes no ano 4 e 5, e terapia combinada de entecavir-lamivudina (seguida por terapia de longo prazo com entecavir) por uma mediana de 20 semanas para 130 de 149 pacientes no ano 3 e por 1 semana para 1 de 121 pacientes no ano 4 no estudo de rollover.
b Inclui pacientes com pelo menos uma medição de HBV DNA em tratamento por PCR em 24 semanas ou até 58 semanas (Ano 1), após 58 semanas até 102 semanas (Ano 2), após 102 semanas até 156 semanas (Ano 3 ), após 156 semanas a 204 semanas (Ano 4) ou após 204 semanas a 252 semanas (Ano 5).
cPacientes que também tiveram substituições LVDr.
aumento ≥ 1 log10 acima do nadir em HBV DNA por PCR, confirmado com medições subsequentes ou no final do ponto de tempo janela.
Substituições de ETVr (além de substituições LVDr rtM204V / I ± rtL180M) foram observadas no início do estudo em isolados de 10/187 (5%) pacientes refratários à lamivudina tratados com entecavir e monitorados para resistência, indicando que o tratamento anterior com lamivudina pode selecionar essas substituições de resistência e que podem existir em baixa frequência antes do tratamento com entecavir. Durante 240 semanas, 3 de 10 pacientes apresentaram rebote virológico (aumento ≥ 1 log10 acima do nadir). O início da resistência ao entecavir em estudos com pacientes refratários à lamivudina durante 240 semanas está resumido na tabela abaixo.
Os resultados refletem o uso de terapia combinada de entecavir-lamivudina (seguida por terapia de longo prazo com entecavir) por uma mediana de 13 semanas para 48 de 80 pacientes no estudo de 3 anos, uma mediana de 38 semanas para 10 de 52 pacientes no estudo de 4 estudo de um ano e de 16 semanas para 1 de 33 pacientes no estudo de rollover de 5 anos.
b Inclui pacientes com pelo menos uma medição de HBV DNA em tratamento por PCR em ou após 24 semanas a 58 semanas (Ano 1), após 58 semanas a 102 semanas (Ano 2), após 102 semanas a 156 semanas (Ano 3), após 156 semanas a 204 semanas (Ano 4) ou após 204 semanas a 256 semanas (Ano 5).
cPacientes que também tiveram substituições LVDr.
aumento ≥ 1 log10 acima do nadir em HBV DNA por PCR, confirmado com medições subsequentes ou no final do ponto de tempo janela.
eEmergência de resistência ao ETV a cada ano; rebote virológico / ano.
Entre os pacientes refratários à lamivudina com DNA do VHB
05.2 "Propriedades farmacocinéticas -
Absorção: o entecavir é rapidamente absorvido com picos de concentração plasmática entre 0,5 e 1,5 horas. A biodisponibilidade absoluta não foi determinada. Com base na excreção urinária do fármaco inalterado, a biodisponibilidade é estimada em pelo menos 70%. Após doses múltiplas de 0,1 a 1 mg, há um aumento proporcional nos valores de Cmax e AUC. O estado estacionário é alcançado entre 6 e 10 dias após a administração de uma vez ao dia com ≈ 2 vezes o tempo de acumulação. Cmax e Cmin no estado estacionário são 4,2 e 0,3 ng / mL para a dose de 0,5 mg e 8,2 e 0,5 ng / mL para a dose de 1 mg, respectivamente. O comprimido e a solução oral foram equivalentes em indivíduos saudáveis; portanto, ambas as formas farmacêuticas podem ser trocadas.
A administração de 0,5 mg de entecavir com uma refeição padrão rica em gordura (945 kcal, 54,6 g de gordura) ou uma refeição leve (379 kcal, 8,2 g de gordura) resultou em um atraso mínimo na absorção (1 - 1,5 horas com o estômago cheio vs 0,75 horas com o estômago vazio), uma diminuição na Cmax de 44 - 46% e uma diminuição na AUC de 18 - 20%. A redução da Cmax e AUC com alimentos não é considerada de relevância clínica em pacientes virgens de nucleosídeos, mas pode afetar a eficácia em pacientes refratários à lamivudina (ver seção 4.2).
Distribuição: O volume de distribuição estimado para o entecavir é superior à água corporal total.Ligação do plasma às proteínas séricas humanas em vitro è ≈ 13%.
Biotransformação: o entecavir não é um substrato, inibidor ou indutor do sistema enzimático CPYP450. Após a administração de 14C-entecavir, não foram observados metabolitos oxidativos ou acetilados e pequenas quantidades de metabolitos de fase II, glucuronido e seus conjugados de sulfato.
Eliminação: o entecavir é eliminado predominantemente pelo rim com recuperação urinária do fármaco inalterado no estado estacionário de aproximadamente 75% da dose. A depuração renal é independente da dose e varia de 360 a 471 mL / min, sugerindo que o entecavir sofre filtração glomerular e secreção tubular distinta. Após atingir os níveis máximos, a concentração plasmática de entecavir diminuiu bi-exponencialmente com uma semivida de eliminação terminal ≥ 128 - 149 horas. O índice de acumulação observado do fármaco é de ≈ 2 vezes com uma dose única diária, sugerindo uma semivida de acumulação efetiva de aproximadamente 24 horas.
Doenças hepáticas: Os parâmetros farmacocinéticos em pacientes com doença hepática moderada a grave são semelhantes aos de pacientes com função hepática normal.
Falência renal: A depuração do entecavir diminui com a diminuição da depuração da creatinina. Numa sessão de hemodiálise de 4 horas, ≈ 13% da dose foi removida e 0,3% foi removida por CAPD. Os dados farmacocinéticos do entecavir após uma dose única de 1 mg (em doentes sem hepatite B crónica infecção) são mostrados na tabela abaixo:
Transplante de fígado: A exposição ao entecavir em pacientes infectados pelo VHB submetidos a transplante de fígado recebendo uma dose estável de ciclosporina A ou tacrolimo (n = 9) foi ≥ 2 vezes a exposição em indivíduos saudáveis com função renal normal. A insuficiência renal contribuiu para o aumento da exposição ao entecavir nestes doentes (ver secção 4.4).
Sexo: A AUC foi 14% maior em mulheres do que em homens, devido a diferenças na função renal e peso. Após ajustes nas diferenças na depuração da creatinina e peso corporal, não houve diferenças na exposição entre homens e mulheres.
Cidadãos idosos: O efeito sobre a idade na farmacocinética do entecavir foi avaliado comparando indivíduos idosos com uma faixa etária entre 65 e 83 anos (idade média em mulheres 69 anos, em homens 74) com indivíduos jovens na faixa etária de 20 e 40 anos (idade média em mulheres 29 anos, em homens 25). A AUC foi 29% maior em idosos do que em jovens, principalmente devido a diferenças na função renal e no peso. Após ajustes para diferenças na depuração da creatinina e peso corporal, os idosos mostraram uma AUC maior. Elevada em 12,5% dos jovens. . A análise farmacocinética da população, incluindo doentes com idades compreendidas entre os 16 e os 75 anos, não demonstrou que a idade afeta significativamente a farmacocinética do entecavir.
Raça: A análise farmacocinética da população não demonstrou que a raça afeta significativamente a farmacocinética do entecavir.No entanto, as conclusões só podem ser tiradas para os grupos caucasianos e asiáticos, uma vez que havia muito poucos indivíduos de outras categorias.
População pediátrica: As fases de equilíbrio farmacocinético do entecavir foram avaliadas (estudo 028) em indivíduos pediátricos HBeAg positivos, com 2 a 18 anos de idade, com doença hepática compensada, dos quais 24 nunca foram tratados com nucleósidos e 19 foram previamente tratados com lamivudina. A exposição ao entecavir em indivíduos virgens de nucleosídeo recebendo uma dose diária de entecavir de 0,015 mg / kg até um máximo de 0,5 mg foi semelhante à exposição alcançada em adultos recebendo uma dose diária de 0,5 mg. A Cmax, AUC (0-24) e Cmin nestes indivíduos foram 6,31 ng / mL, 18,33 ng h / mL e 0,28 ng / mL, respectivamente.
A exposição ao entecavir em indivíduos previamente tratados com lamivudina e recebendo uma dose diária de entecavir de 0,030 mg / kg até uma dose máxima de 1,0 mg foi semelhante à exposição alcançada em adultos recebendo uma dose diária de 1,0 mg. A Cmax, AUC (0-24) e Cmin nestes indivíduos foram 14,48 ng / mL, 38,58 ng h / mL e 0,47 ng / mL, respectivamente.
05.3 Dados de segurança pré-clínica -
Em estudos de toxicologia de dose repetida em cães, "foi observada inflamação perivascular reversível do sistema nervoso central. No entanto, essa inflamação não foi detectada em doses 9 e 10 vezes maiores do que em humanos (quando administrando doses de 0,5 e 1 mg). Este efeito não ocorreu em estudos de dose repetida em outras espécies, incluindo macacos tratados com entecavir diariamente durante 1 ano em doses ≥ 100 vezes as doses administradas em humanos.
Em estudos de toxicologia reprodutiva nos quais os animais receberam entecavir durante 4 semanas, não foi observada perda de fertilidade em ratos machos ou fêmeas com doses elevadas. Alterações nos testículos (degeneração tubular seminífera) foram observadas em estudos de toxicologia de dose repetida em roedores e cães em doses ≥ 26 vezes as doses administradas em humanos. Mudanças nos testículos não foram observadas em um estudo de 1 ano em macacos. Em ratas e coelhas grávidas administradas com entecavir, nenhum nível real de embriotoxicidade ou toxicidade materna correspondeu a doses ≥ 21 vezes as doses administradas em humanos. Em altas doses, os seguintes efeitos foram observados em ratos: toxicidade materna, toxicidade embriofetal (reabsorção), reduções no peso corporal fetal,
cauda e vértebras, ossificação reduzida (das vértebras, esternebras e falanges) e vértebras extra lombares. Em doses elevadas, foram observados os seguintes efeitos em coelhos: toxicidade embriofetal (reabsorção), ossificação reduzida (do osso hióide), um aumento nos casos da costela 13. Num estudo peri-pós-natal em ratos, não foi Não foram observados eventos adversos na prole.Num estudo separado no qual o entecavir foi administrado na dose de 10 mg / kg a ratas grávidas e lactantes, foram observados tanto a exposição fetal ao entecavir quanto a secreção de entecavir no leite. Em ratos juvenis tratados com entecavir do quarto ao octogésimo dia pós-natal, uma resposta moderadamente reduzida a estímulos acústicos foi observada durante o período de recuperação (110-114 dias pós-natal), mas não durante o período de administração com valores de AUC ≥ 92 vezes maiores do que aqueles em humanos em uma dose de 0,5 mg ou uma dose pediátrica equivalente. Dada a margem de exposição, este achado é considerado de improvável relevância clínica.
A genotoxicidade não foi observada com o teste de mutagenicidade microbiana de Ames, o teste de mutação do gene de células de mamíferos ou o teste de transformação de células embrionárias de hamster sírio. Tanto um estudo de micronúcleo quanto um estudo de reparo de DNA em ratos foram negativos. O entecavir foi clastogênico em culturas de linfócitos humanos em concentrações substancialmente mais altas do que as alcançadas no ambiente clínico.
Nos estudos de carcinogenicidade de dois anos: em camundongos machos, um aumento nos casos de câncer de pulmão com doses ≥ 4 e ≥ 2 vezes as doses em humanos com doses de 0,5 mg e 1 mg, respectivamente. O desenvolvimento do tumor foi precedido pela proliferação de pneumócitos no pulmão, o que, entretanto, não foi observado em ratos, cães ou macacos, sugerindo que o evento chave no desenvolvimento do câncer de pulmão em camundongos é provavelmente específico da espécie. Os seguintes efeitos foram observado com a administração por longos períodos de tempo: um aumento nos casos de outros tipos de tumores, incluindo glioma cerebral em ratos machos e fêmeas, câncer de fígado em ratos machos, tumores vasculares benignos em ratos fêmeas e adenomas e carcinomas do fígado em ratos fêmeas. No entanto, nenhum efeito preciso sobre os níveis pode ser estabelecido.A previsibilidade de tais observações em humanos é desconhecida.
06.0 INFORMAÇÕES FARMACÊUTICAS -
06.1 Excipientes -
Núcleo do tablet:
crospovidona
lactose monohidratada
estearato de magnesio
celulose microcristalina
povidona
Revestimento do comprimido:
dióxido de titânio
hipromelose
macrogol 400
Polissorbato 80 (E433)
06.2 Incompatibilidade "-
Não é relevante.
06.3 Período de validade "-
2 anos
06.4 Precauções especiais de armazenamento -
Bolha:
Não armazene acima de 30 ° C. Armazene na caixa original.
Garrafas:
Não armazene acima de 25 ° C. Mantenha o frasco bem fechado.
06.5 Natureza da embalagem primária e conteúdo da embalagem -
Cada caixa contém:
§ 30 x 1 comprimido revestido por película; 3 blisters de 10 x 1 comprimido revestido por película, cada blister de dose unitária Alu / Alu perfurado ou:
§ 90 x 1 comprimido revestido por película; 9 blisters de 10 x 1 comprimido revestido por película, cada blister de unidade de dose Alu / Alu perfurado
Frasco de polietileno de alta densidade (HDPE) com fecho de polipropileno resistente à abertura por crianças, contendo 30 comprimidos revestidos por película. Cada caixa contém uma garrafa.
Nem todos os tamanhos de embalagens e tipos de recipientes podem ser comercializados.
06.6 Instruções de uso e manuseio -
O medicamento não utilizado e os resíduos derivados deste medicamento devem ser eliminados de acordo com os regulamentos locais.
07.0 TITULAR DA "AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO" -
BRISTOL-MYERS SQUIBB PHARMA EEIG
Uxbridge Business Park
Sanderson Road
Uxbridge UB8 1DH
Reino Unido
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO -
Bolha: EU / 1/06/343/003
037221076
EU / 1/06/343/006
Garrafa: EU / 1/06/343/001
037221052
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO -
Data da primeira autorização: 26 de junho de 2006
Data da última renovação: 26 de junho de 2011
10.0 DATA DE REVISÃO DO TEXTO -
D.CCE agosto de 2014