Ingredientes ativos: Esomeprazol
LUCEN 20 mg comprimidos gastrorresistentes
LUCEN 40 mg comprimidos gastrorresistentes
As bulas Lucen estão disponíveis para os tamanhos de embalagem: - LUCEN 20 mg comprimidos gastrorresistentes, LUCEN 40 mg comprimidos gastrorresistentes
- LUCEN 10 mg grânulos gastrorresistentes para suspensão oral, em saqueta
- LUCEN 40 mg pó para solução injetável / infusão
Indicações Por que o Lucen é usado? Para que serve?
LUCEN contém um medicamento denominado esomeprazol. Pertence a um grupo de medicamentos denominados “inibidores da bomba de protões”, que atuam reduzindo a quantidade de ácido produzida pelo estômago.
LUCEN é usado para o tratamento das seguintes doenças:
- "Doença do refluxo gastroesofágico" (DRGE). Ocorre quando o ácido do estômago escapa para o esôfago (o tubo que conecta a garganta ao estômago), causando dor, inflamação e queimação.
- Úlceras do estômago ou do intestino superior infectadas com uma bactéria chamada "Helicobacter pylori". Se você tiver essas doenças, o médico também pode prescrever antibióticos para tratar a infecção e permitir que a úlcera cicatrize.
- Úlceras estomacais causadas por medicamentos denominados AINEs (Anti-Inflamatórios Não Esteróides). LUCEN também pode ser usado para prevenir a formação de úlceras estomacais durante o tratamento com AINEs.
- Excesso de ácido estomacal causado por tumor no pâncreas (síndrome de Zollinger-Ellisson).
- Tratamento prolongado de ressangramento de úlceras, após prevenção com administração intravenosa de Lucen
Contra-indicações Quando Lucen não deve ser usado
Não tome LUCEN:
- se tem alergia (hipersensibilidade) ao esomeprazol ou a qualquer outro componente deste medicamento (listados na seção: Outras informações).
- se você é alérgico a outros medicamentos inibidores da bomba de prótons (por exemplo, pantoprazol, lansoprazol, rabeprazol, omeprazol).
- se está a tomar um medicamento contendo nelfinavir (usado para tratar o VIH).
Você não deve tomar LUCEN se ele se enquadrar em algum dos casos acima. Em caso de dúvida, consulte o seu médico ou farmacêutico antes de tomar LUCEN.
Precauções de uso O que você precisa saber antes de tomar Lucen
Tome especial cuidado com LUCEN
Fale com o seu médico ou farmacêutico antes de tomar LUCEN se:
- Você tem problemas graves de fígado
- Você tem problemas renais graves.
LUCEN pode mascarar os sintomas de outras doenças. Portanto, se alguma das situações a seguir acontecer com você antes de começar a tomar ou enquanto estiver tomando LUCEN, informe o seu médico imediatamente:
- Você perde muito peso sem motivo ou tem problemas para engolir
- Ocorre dor de estômago ou indigestão
- Comece a vomitar comida ou sangue
- As fezes são pretas (fezes com manchas de sangue).
Se LUCEN lhe foi prescrito "conforme necessário", entre em contato com o seu médico se os sintomas persistirem ou se alterarem as características.
Se você tomar um inibidor da bomba de prótons como LUCEN, especialmente por mais de um ano, você pode ter um risco ligeiramente aumentado de fratura do quadril, punho ou coluna vertebral. Se você tem osteoporose ou está tomando corticosteroides (o que pode aumentar o risco de osteoporose) consulte o seu médico
Interações Quais medicamentos ou alimentos podem alterar o efeito do Lucen
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica.
Na verdade, LUCEN pode afetar a forma como alguns medicamentos atuam e alguns medicamentos podem ter um efeito sobre LUCEN.Não deve tomar LUCEN se estiver a tomar um medicamento contendo nelfinavir (usado para tratar o VIH).
Informe o seu médico ou farmacêutico se estiver tomando algum dos seguintes medicamentos:
- Atazanavir (usado para tratar o HIV)
- Clopidogrel (usado para prevenir coágulos sanguíneos)
- Cetoconazol, itraconazol ou voriconazol (usados para tratar infecções causadas por fungos).
- Erlotinib (usado para tratar o câncer).
- Citalopram, imipramina ou clomipramina (usados para tratar a depressão).
- Diazepam (usado no tratamento da ansiedade, no relaxamento muscular ou na epilepsia).
- Fenitoína (usada na epilepsia) Se estiver a tomar fenitoína, o seu médico terá de o monitorizar quando iniciar ou interromper o tratamento com LUCEN.
- Medicamentos usados para tornar o sangue mais fluido, como a varfarina. O seu médico pode monitorizá-lo quando iniciar ou parar o tratamento com LUCEN.
- Cilostazol (usado para tratar claudicação intermitente - dor nas pernas ao caminhar devido ao fornecimento insuficiente de sangue).
- Cisaprida (usado para indigestão e azia).
- Digoxina (usada para problemas cardíacos).
- Metotrexato (um medicamento de quimioterapia usado em altas doses para tratar o câncer) - se você estiver tomando altas doses de metotrexato, o seu médico pode interromper temporariamente o seu tratamento com Lucen.
- Tacrolimus (usado em transplantes de órgãos)
- Rifampicina (usada para tratar a tuberculose).
- Erva de São João (Hypericum perforatum) (usada para tratar a depressão).
Se o seu médico prescreveu antibióticos como amoxicilina e claritromicina com LUCEN para o tratamento de úlceras causadas por infecção por Helicobacter pylori, é muito importante que informe o seu médico sobre os seus outros medicamentos.
Avisos É importante saber que:
Gravidez e amamentação
Antes de tomar LUCEN, informe o seu médico se estiver grávida ou se quiser engravidar. Consulte o seu médico ou farmacêutico antes de tomar qualquer medicamento. O seu médico decidirá se você pode tomar LUCEN durante este período.
Não se sabe se LUCEN passa para o leite materno, pelo que não deve tomar LUCEN se estiver a amamentar.
Tomar LUCEN com alimentos e bebidas
Os comprimidos podem ser tomados com o estômago cheio ou vazio.
Condução e utilização de máquinas
É improvável que LUCEN afete a sua capacidade de conduzir ou usar quaisquer ferramentas ou máquinas.
Informações importantes sobre alguns ingredientes de LUCEN
Os comprimidos gastrorresistentes LUCEN contêm sacarose, que é um tipo de açúcar. Se o seu médico lhe disse que tem "intolerância a alguns açúcares, consulte-o antes de tomar o medicamento.
Dosagem e método de uso Como usar Lucen: Dosagem
Tome LUCEN sempre de acordo com as indicações do médico. Em caso de dúvida, consulte o seu médico ou farmacêutico.
- Os comprimidos gastrorresistentes LUCEN não são recomendados para crianças com menos de 12 anos de idade
- Se está a tomar este medicamento há muito tempo, o seu médico irá monitorizá-lo (particularmente se estiver a tomar o medicamento há mais de um ano)
- Se o seu médico lhe disse para tomar o medicamento quando necessário, conforme necessário, informe o seu médico se os seus sintomas mudarem.
Tomando o remédio
- Você pode tomar os comprimidos a qualquer hora do dia.
- Pode tomar os comprimidos com o estômago cheio ou vazio.
- Engula os comprimidos inteiros com um copo de água. Não mastigue ou esmague os comprimidos porque contêm grânulos revestidos que protegem o medicamento da acidez gástrica, pelo que é importante não danificar os grânulos.
O que fazer se você tiver problemas para engolir os comprimidos
Se você tiver problemas para engolir os comprimidos:
- Coloque os comprimidos em um copo de água sem gás. Outros líquidos não devem ser usados
- Mexa até os comprimidos se dissolverem (a mistura não terá uma aparência límpida). Beba imediatamente ou pelo menos dentro de 30 minutos. Sempre misture antes de beber
- Para se certificar de que tomou todo o medicamento, enxagúe bem o copo enchendo-o até meio com água e beba.As partículas sólidas contêm o medicamento e não devem ser mastigadas ou esmagadas.
Se não conseguir engolir, o comprimido pode ser misturado com um pouco de água, inserido numa seringa e administrado através de um tubo diretamente no estômago (tubo gástrico).
Quanta medicação tomar
- O seu médico irá aconselhá-lo sobre o número de comprimidos a tomar e durante quanto tempo. Esta é uma função da sua condição física, idade e condição hepática.
- As doses usuais são fornecidas abaixo.
Tratamento da azia causada pela doença do refluxo gastroesofágico (DRGE):
Adultos e crianças a partir dos 12 anos:
- Se o seu médico constatou que o seu esôfago está ligeiramente danificado, a dose habitual é um comprimido gastrorresistente de LUCEN de 40 mg uma vez por dia durante 4 semanas. O seu médico pode dizer-lhe para continuar o tratamento, tomando a mesma dose, por mais 4 semanas, caso o esófago não tenha cicatrizado.
- Após a cicatrização do esófago, a dose habitual é um comprimido gastrorresistente de LUCEN de 20 mg uma vez por dia.
- Se o esófago não estiver danificado, a dose habitual é um comprimido gastro-resistente de LUCEN 20 mg por dia. Quando os seus sintomas estiverem controlados, o seu médico irá informá-lo de que pode tomar o medicamento quando necessário, até ao máximo de um comprimido gastro-resistente -comprimido resistente de Lucen 20 mg por dia.
- Se tiver problemas graves de fígado, o seu médico irá dar-lhe uma dose mais baixa.
Tratamento de úlceras causadas por infecção por Helicobacter pylori e prevenção do seu reaparecimento:
- Adultos a partir dos 18 anos de idade: a dose habitual é um comprimido gastrorresistente de LUCEN 20 mg duas vezes por dia durante uma semana.
- O seu médico também lhe dirá para tomar antibióticos chamados amoxicilina e claritromicina.
Tratamento de úlceras gástricas causadas por AINEs (medicamentos anti-inflamatórios não esteroidais):
- Adultos a partir dos 18 anos de idade: a dose habitual é um comprimido gastrorresistente de LUCEN 20 mg uma vez por dia durante 4 a 8 semanas.
Prevenção de úlceras estomacais se você estiver tomando AINEs (medicamentos anti-inflamatórios não esteroidais):
- Adultos a partir dos 18 anos de idade: a dose habitual é um comprimido gastrorresistente de LUCEN 20 mg uma vez por dia.
Tratamento do excesso de ácido gástrico causado por um crescimento no pâncreas (síndrome de Zollinger-Ellison):
- Adultos a partir dos 18 anos de idade: a dose habitual é um comprimido gastrorresistente de LUCEN 40 mg duas vezes por dia.
- O seu médico ajustará a dose de acordo com a sua necessidade e também decidirá por quanto tempo continuar o tratamento.
A dose máxima é de 80 mg duas vezes ao dia.
Tratamento prolongado de ressangramento de úlceras, após prevenção com administração intravenosa de Lucen:
A dose habitual é um comprimido de Lucen 40 mg uma vez por dia durante 4 semanas.
Overdose O que fazer se você tiver tomado muito Lucen
Se você tomar mais LUCEN do que deveria
Se tomou mais LUCEN do que o prescrito pelo seu médico, informe o seu médico ou farmacêutico imediatamente.
Se você esquecer de tomar LUCEN
- Se você se esqueceu de tomar uma dose de LUCEN, tome-a assim que se lembrar. Se estiver quase na hora da próxima dose, pule a dose esquecida.
- Não tome uma dose dupla (duas doses simultaneamente) para compensar uma dose esquecida.
Efeitos colaterais Quais são os efeitos colaterais do Lucen
Como todos os medicamentos, LUCEN pode causar efeitos secundários, embora nem todas as pessoas os tenham.
Se você notar algum dos seguintes efeitos colaterais graves, pare de tomar LUCEN e entre em contato com o seu médico imediatamente:
- Sibilos repentinos, inchaço dos lábios, língua e garganta ou corpo, erupção na pele, desmaios ou dificuldade em engolir (reação alérgica grave).
- Vermelhidão da pele com bolhas ou descamação. Bolhas graves e sangramento também podem aparecer nos lábios, olhos, boca, nariz e órgãos genitais. Pode ser "síndrome de Stevens-Johnson" ou "necrólise epidérmica tóxica".
- Pele amarela, urina escura e cansaço podem ser sintomas de problemas de fígado. Estes efeitos são raros, afetando menos de 1 em 1000 pessoas.
Outros efeitos colaterais incluem:
Comum (afeta menos de 1 em 10 pessoas):
- Dor de cabeça.
- Efeitos no estômago ou intestinos: diarreia, dor de estômago, prisão de ventre, flatulência.
- Náusea ou vômito.
Pouco frequentes (afetam menos de 1 em 100 pessoas):
- Inchaço nos pés e tornozelos.
- Sono perturbado (insônia).
- Tontura, alfinetes e agulhas, sonolência.
- Tontura.
- Boca seca
- Alterações nas análises ao sangue que verificam o funcionamento do fígado.
- Erupção cutânea, urticária e coceira.
- Fratura do quadril, punho ou coluna (se Lucen for usado em altas doses e por períodos prolongados).
Raros (afetam menos de 1 em 1.000 pessoas):
- Problemas sanguíneos, como redução do número de glóbulos brancos e plaquetas. Isso pode causar fraqueza, hematomas ou facilitar as infecções.
- Níveis baixos de sódio no sangue. Isso pode causar fraqueza, vômitos e cólicas.
- Sentir-se agitado, confuso ou deprimido.
- Mudanças de gosto.
- Problemas de visão, como visão turva.
- Chiado repentino ou falta de ar (broncoespasmo).
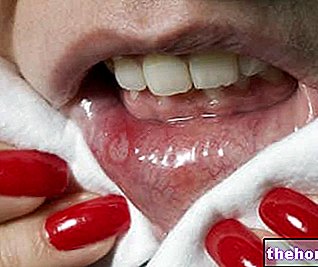
- Inflamação do interior da boca.
- Infecção chamada "sapinho", que pode afetar o intestino e é causada por um fungo.
- Problemas de fígado, incluindo icterícia, que pode causar pele amarelada, urina escura e cansaço.
- Perda de cabelo (alopecia).
- Erupção cutânea por exposição ao sol.
- Dor nas articulações (artralgia) ou dor muscular (mialgia).
- Sensação geral de indisposição e falta de força.
- Aumento da transpiração.
Muito raro (afeta menos de 1 em 10.000 pessoas):
- Alterações no número de células sanguíneas, incluindo agranulocitose (falta de células brancas do sangue).
- Agressão.
- Ver, sentir ou ouvir coisas que não existem (alucinações).
- Problemas graves do fígado que levam à insuficiência hepática e inflamação do cérebro.
- Início súbito de erupção cutânea grave ou formação de bolhas ou descamação da pele. Isso pode estar associado a febre alta e dor nas articulações (eritema multiforme, síndrome de Stevens-Johnson, necrólise epidérmica tóxica).
- Fraqueza muscular.
- Problemas renais graves.
- Aumento dos seios em homens.
Desconhecido (a frequência não pode ser estimada a partir dos dados disponíveis)
- Se você tomar LUCEN por mais de três meses, seus níveis de magnésio no sangue podem cair. Níveis baixos de magnésio podem se manifestar com fadiga, contrações musculares involuntárias, desorientação, convulsões, tonturas, aumento da freqüência cardíaca. Se você tiver algum desses sintomas, consulte o seu médico imediatamente. Níveis baixos de magnésio também podem levar à redução dos níveis de potássio ou cálcio no sangue. O seu médico deve decidir se deve verificar os seus níveis de magnésio no sangue periodicamente.
- Inflamação no intestino (levando à diarreia).
LUCEN pode, em casos muito raros, afetar os glóbulos brancos, levando à imunodeficiência. Se você tiver uma infecção com sintomas como febre com grave deterioração de sua condição física geral ou febre com sintomas de infecção local, como dor no pescoço, garganta ou boca ou dificuldade para urinar, você deve consultar o seu médico o mais rápido possível. essa falta de glóbulos brancos (agranulocitose) pode ser excluída por meio de um exame de sangue. É importante que você forneça informações sobre os medicamentos que está tomando. Não se preocupe com a lista de possíveis efeitos secundários acima. Pode não ter nenhum. Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico ou farmacêutico.
Expiração e retenção
- Manter fora do alcance e da vista das crianças.
- Não armazene acima de 30 ° C.
- Conservar na embalagem de origem (blister) ou manter o recipiente bem fechado (frasco) para proteger da umidade.
- Não utilize os comprimidos após o prazo de validade (VAL) impresso na embalagem, carteira ou blister. A data de validade refere-se ao último dia do mês.
- Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Isto ajudará a proteger o ambiente.
OUTRA INFORMAÇÃO
O que LUCEN contém
O ingrediente ativo é o esomeprazol. Os comprimidos gastrorresistentes LUCEN estão presentes em 2 dosagens contendo 20 ou 40 mg de esomeprazol (como tri-hidrato de magnésio).
Os outros ingredientes são: monoestearato de glicerol 40-55, hiprolose, hipromelose, óxido de ferro (vermelho-castanho, amarelo) (E172, apenas para comprimidos de 20 mg), estearato de magnésio, copolímero de ácido metacrílico, etil acrilato de dispersão (1: 1) a 30 %, celulose microcristalina, parafina sintética, macrogóis, polissorbato 80, crospovidona, estearil fumarato de sódio, esferas de sacarose (sacarose e amido de milho), talco, dióxido de titânio (E171), citrato de trietila.
Descrição da aparência de LUCEN e conteúdo da embalagem
- Os comprimidos gastrorresistentes de LUCEN 20 mg são rosa claro com A / EH de um lado e 20 mg do outro.
- Os comprimidos gastrorresistentes de LUCEN 40 mg são cor-de-rosa com A / EI numa das faces e 40 mg na outra.
- Os comprimidos estão em embalagens blister, carteiras e / ou frascos contendo
- 20 mg, 40 mg: frasco de comprimidos de 2-5-7-14-15-28-30-56-60-100-140 (28x5).
- 20 mg, 40 mg: blister ou carteira de 3-7-7x1-14-15-25x1-28-30-50x1- 56-60-90-98-100x1-140 comprimidos.
Nem todos os tamanhos de embalagem podem ser comercializados
Folheto Informativo Fonte: AIFA (Agência Italiana de Medicamentos). Conteúdo publicado em janeiro de 2016. As informações apresentadas podem não estar atualizadas.
Para ter acesso à versão mais atualizada, é aconselhável acessar o site da AIFA (Agência Italiana de Medicamentos). Isenção de responsabilidade e informações úteis.
01.0 NOME DO MEDICAMENTO
SOLUÇÃO LUCENTIS 10 MG / ML PARA INJEÇÃO
02.0 COMPOSIÇÃO QUALITATIVA E QUANTITATIVA
Um ml contém 10 mg de ranibizumab *. Cada frasco para injetáveis contém 2,3 mg de ranibizumab em 0,23 ml de solução.
* Ranibizumab é um fragmento de anticorpo monoclonal humanizado produzido nas células de Escherichia coli por tecnologia de DNA recombinante.
Para a lista completa de excipientes, consulte a seção 6.1.
03.0 FORMA FARMACÊUTICA
Solução injetável
Solução aquosa límpida, incolor a amarelo pálido.
04.0 INFORMAÇÕES CLÍNICAS
04.1 Indicações terapêuticas
Lucentis é indicado em adultos para:
• Tratamento da degeneração macular neovascular (úmida) relacionada à idade (DMRI)
• Tratamento da deficiência visual causada por edema macular diabético (DME)
• O tratamento da deficiência visual causada por "edema macular secundário à oclusão da veia retiniana (RVO RVO ou RVO central)
• Tratamento da deficiência visual causada por neovascularização coroidal (CNV) secundária à miopia patológica (PM)
04.2 Posologia e método de administração
Lucentis deve ser administrado por um oftalmologista qualificado com experiência em injeções intravítreas.
Posologia para o tratamento da DMRI úmida
A dose recomendada de Lucentis é de 0,5 mg administrada mensalmente como uma injeção intravítrea única. Isso corresponde a um volume injetado de 0,05 ml.
O tratamento é administrado mensalmente e continuado até que a acuidade visual máxima seja atingida, ou seja, a acuidade visual do paciente está estável por três verificações mensais consecutivas realizadas durante o tratamento com ranibizumabe.
Portanto, a acuidade visual dos pacientes deve ser monitorada mensalmente.
O tratamento deve ser retomado quando a monitoração indicar uma diminuição na acuidade visual devido à DMRI úmida. As injeções mensais devem ser administradas até que a acuidade visual estável seja alcançada novamente por três verificações mensais consecutivas (isto implica um mínimo de duas injeções). O intervalo entre duas doses não deve ser inferior a um mês.
Posologia para o tratamento da deficiência visual causada por DME ou edema macular secundário a RVO
A dose recomendada de Lucentis é de 0,5 mg administrada mensalmente como uma injeção intravítrea única. Isso corresponde a um volume injetado de 0,05 ml.
O tratamento é administrado mensalmente e continuado até que a acuidade visual máxima seja atingida, ou seja, a acuidade visual do paciente está estável por três verificações mensais consecutivas realizadas durante o tratamento com ranibizumabe. Se não houver melhora na acuidade visual durante o período das três primeiras injeções, a continuação do tratamento não é recomendada.
Portanto, a acuidade visual dos pacientes deve ser monitorada mensalmente.
O tratamento deve ser retomado quando a monitoração indicar diminuição da acuidade visual devido a EMD ou edema macular secundário a RVO. As injeções mensais devem ser administradas até que a acuidade visual estável seja alcançada novamente por três verificações mensais consecutivas (isso envolve um mínimo de duas injeções). O intervalo entre as duas doses não deve ser inferior a um mês.
Lucentis e fotocoagulação a laser em DME e edema macular secundário a BRVO
Existe alguma experiência de administração de Lucentis concomitantemente com fotocoagulação a laser (ver secção 5.1). Quando administrado no mesmo dia, Lucentis deve ser administrado pelo menos 30 minutos após a fotocoagulação a laser. Lucentis pode ser administrado a pacientes que já receberam fotocoagulação a laser.
Posologia para o tratamento da deficiência visual causada por CNV secundária a PM
O tratamento deve ser iniciado com uma única injeção.
Se o monitoramento revelar sinais de atividade da doença, como acuidade visual reduzida e / ou sinais de lesão, o tratamento adicional é recomendado.
O monitoramento da doença pode incluir um exame clínico, tomografia de coerência óptica (OCT) ou angiografia com fluoresceína (AF).
Embora alguns doentes possam necessitar apenas de uma ou duas injecções durante o primeiro ano de tratamento, alguns podem necessitar de um tratamento mais frequente (ver secção 5.1). Portanto, o monitoramento mensal é recomendado durante os primeiros dois meses e pelo menos a cada três meses durante o primeiro ano de tratamento. Após o primeiro ano, a frequência do acompanhamento pode ser determinada pelo médico.
O intervalo entre duas doses não deve ser inferior a um mês.
Lucentis e terapia fotodinâmica com Visudyne em CNV secundária a PM
Não há experiência com a administração de Lucentis em combinação com Visudyne.
Populações especiais
Insuficiência Hepática
Lucentis não foi estudado em pacientes com insuficiência hepática. No entanto, nenhuma consideração especial é necessária para este polação.
Falência renal
Não é necessário ajuste de dose em doentes com insuficiência renal (ver secção 5.2).
Cidadãos idosos
Não é necessário ajuste de dose em idosos. A experiência em pacientes com EMD com mais de 75 anos é "limitada".
População pediátrica
A segurança e eficácia de Lucentis em crianças e adolescentes com idade inferior a 18 anos não foram estabelecidas.Não existem dados disponíveis.
Método de administração
Frascos para injectáveis de uso único apenas para uso intravítreo.
Antes da administração, Lucentis deve ser verificado visualmente quanto à presença de partículas e descoloração.
O procedimento de injeção deve ser realizado em condições assépticas, que incluem desinfecção das mãos como em qualquer procedimento cirúrgico, luvas estéreis, campo estéril e blefarostato estéril (ou equivalente) e possibilidade de realização de paracentese estéril (se necessário A história do paciente de reações de hipersensibilidade deve ser avaliada cuidadosamente antes do procedimento intravítreo (ver seção 4.4). Anestesia adequada e um antimicrobiano tópico de amplo espectro devem ser administrados antes da injeção para desinfetar a superfície periocular, ocular e palpebral, de acordo com a prática clínica.
Para obter informações sobre a preparação de Lucentis, consulte a seção 6.6.
Insira a agulha de injeção 3,5-4,0 mm posterior ao limbo, na câmara vítrea, evitando o meridiano horizontal e direcionando a agulha em direção ao centro do globo ocular. Injete o volume de injeção de 0,05 ml; mude o local da esclera para as injeções subsequentes.
04.3 Contra-indicações
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes listados na seção 6.1.
Pacientes com infecções oculares ou perioculares atuais ou suspeitas.
Pacientes com inflamação intraocular grave em andamento.
04.4 Advertências especiais e precauções adequadas de uso
Reações relacionadas à injeção intravítrea
As injecções intravítreas, incluindo aquelas com Lucentis, foram associadas a endoftalmite, inflamação intraocular, descolamento regmatogénico da retina, ruptura da retina e catarata traumática iatrogénica (ver secção 4.8). Devem sempre ser utilizadas técnicas de injeção asséptica adequadas para a administração de Lucentis. Além disso, os pacientes devem ser monitorados na semana seguinte à injeção para permitir um tratamento rápido em caso de infecção. Os pacientes devem ser instruídos sobre como relatar quaisquer sintomas sugestivos de endoftalmite ou qualquer um dos eventos acima sem demora.
Aumentos na pressão intraocular
Foram observados aumentos transitórios da pressão intraocular (PIO) 60 minutos após a injeção de Lucentis. Também foram observados aumentos prolongados da PIO (ver secção 4.8). A pressão intraocular e a perfusão da cabeça do nervo óptico devem ser monitorizados e tratados de forma adequada.
Tratamento bilateral
Os dados limitados sobre o uso bilateral de Lucentis (incluindo dosagem no mesmo dia) não indicam um risco aumentado de eventos adversos sistêmicos em comparação com o tratamento unilateral.
Imunogenicidade
Existe um potencial de imunogenicidade com Lucentis. Como existe a possibilidade de aumento da exposição sistêmica em indivíduos com DME, um risco aumentado de desenvolver hipersensibilidade nesta população de pacientes não pode ser excluído. Os pacientes também devem ser educados sobre como relatar se a inflamação intraocular piorar, porque pode ser um sintoma clínico atribuível a a formação de anticorpos intraoculares.
Uso concomitante com outros anti-VEGFs (fator de crescimento endotelial vascular)
Lucentis não deve ser administrado concomitantemente com outros medicamentos anti-VEGF (sistémicos ou oculares).
Descontinuação de Lucentis
A dose não deve ser administrada e o tratamento não deve ser retomado antes do próximo tratamento programado no caso de:
• uma diminuição na melhor acuidade visual corrigida (BCVA) ≥30 letras em comparação com a última avaliação;
• uma pressão intraocular ≥30 mmHg;
• uma ruptura da retina;
• uma "hemorragia sub-retiniana que se estende até o centro da fóvea ou se a extensão da hemorragia for ≥50% da área total da lesão";
• cirurgia intraocular realizada ou planejada nos 28 dias anteriores ou nos próximos.
Ruptura do epitélio pigmentar da retina
Os fatores de risco associados ao início da ruptura epitelial do pigmento da retina após terapia anti-VEGF para DMRI úmida incluem grande e / ou alto descolamento do epitélio do pigmento da retina. Ao iniciar a terapia com Lucentis, deve-se ter cuidado em pacientes com esses fatores de risco de ruptura do epitélio pigmentar da retina.
Descolamento de retina regmatogênico ou orifícios maculares
O tratamento deve ser interrompido em indivíduos com descolamento retiniano regmatogênico ou orifícios maculares de estágio 3 ou 4.
Populações com dados limitados
Existe apenas experiência limitada no tratamento de indivíduos com DME secundária à diabetes tipo I. Lucentis não foi estudado em pacientes que haviam recebido injeções intravítreas, em pacientes com infecções sistêmicas ativas, retinopatia diabética proliferativa ou em pacientes com condições médicas concomitantes .como descolamento da retina ou buraco macular .Também não existe experiência de tratamento com Lucentis em doentes diabéticos com HbAlc superior a 12% e hipertensão não controlada. A falta de informação deve ser considerada pelo médico ao tratar estes pacientes.
Em pacientes com PM, existem dados limitados sobre o efeito de Lucentis em pacientes previamente tratados com terapia fotodinâmica malsucedida com verteporfina (vPDT). Além disso, embora um efeito consistente tenha sido observado em indivíduos com lesões subfoveais e justtafoveais, não há dados suficientes sobre o efeito de Lucentis em indivíduos com PM com lesões extrafoveais.
Efeitos sistêmicos após administração intravítrea
Eventos adversos sistêmicos, incluindo hemorragias não oculares e eventos tromboembólicos arteriais, foram relatados após injeção intravítrea de inibidores de VEGF.
Existem dados limitados sobre a segurança do tratamento de EMD, edema macular causado por RVO e CNV secundário a PM em doentes com história de AVC ou ataques isquémicos transitórios. Deve ter-se especial cuidado ao tratar estes doentes (ver secção 4.8).
Episódios anteriores de RVO, ramo isquêmico e RVO central
A experiência no tratamento de pacientes com episódios anteriores de RVO e de pacientes com RVO de ramo isquêmico (BRVO) e RVO central (CRVO) é limitada. Em pacientes com RVO que apresentam perda da função visual com sinais clínicos de isquemia irreversível, tratamento não é recomendado.
04.5 Interações com outros medicamentos e outras formas de interação
Não foram realizados estudos convencionais de interação.
Para o uso combinado de terapia fotodinâmica (PDT) com verteporfina e Lucentis na DMRI úmida e PM, ver seção 5.1.
Para o uso combinado de fotocoagulação a laser e Lucentis no tratamento de DME e BRVO, ver seções 4.2 e 5.1.
04.6 Gravidez e lactação
Mulheres com potencial para engravidar / contracepção em mulheres
Mulheres com potencial para engravidar devem usar métodos contraceptivos eficazes durante o tratamento.
Gravidez
Para o ranibizumab, não existem dados clínicos disponíveis em gravidezes expostas. Os estudos em macacos cynomolgus não mostraram efeitos nefastos diretos ou indiretos no que diz respeito à gravidez ou ao desenvolvimento embrionário / fetal (ver secção 5.3). A exposição sistêmica ao ranibizumabe é baixa após a administração ocular, mas devido ao mecanismo de ação, o ranibizumabe deve ser considerado como potencialmente teratogênico e embrionário / fetotóxico. Portanto, o ranibizumabe não deve ser usado durante a gravidez, a menos que os benefícios esperados superem o risco potencial para o feto. Mulheres que planejam engravidar e foram tratadas com ranibizumabe devem esperar pelo menos 3 meses após a última dose de ranibizumabe antes de conceber um bebê.
Gravidez
Não se sabe se Lucentis é excretado no leite humano. É recomendado não amamentar enquanto estiver usando Lucentis.
Fertilidade
Não existem dados disponíveis sobre fertilidade.
04.7 Efeitos sobre a capacidade de dirigir e usar máquinas
O procedimento de tratamento com Lucentis pode induzir distúrbios visuais transitórios que podem afetar a capacidade de conduzir ou utilizar máquinas (ver secção 4.8). Os pacientes que apresentarem esses sintomas não devem dirigir ou operar máquinas até que esses distúrbios visuais transitórios cessem.
04.8 Efeitos indesejáveis
Resumo do perfil de segurança
A maioria das reações adversas notificadas após a administração de Lucentis estão relacionadas com o procedimento de injeção intravítrea.
As reações adversas oculares notificadas com mais frequência após a injeção de Lucentis são: dor ocular, hiperemia ocular, aumento da pressão intraocular, vitreíte, descolamento do vítreo, hemorragia retinal, distúrbio visual, moscas volantes (moscas volantes), hemorragia conjuntival, irritação ocular, sensação de corpo estranho no olho, lacrimejamento aumentado, blefarite, olho seco e olho com coceira.
As reações adversas não oculares notificadas com mais frequência são cefaleia, nasofaringite e artralgia.
As reações adversas notificadas com menos frequência, mas mais graves incluem endoftalmite, cegueira, descolamento da retina, ruptura da retina e catarata traumática iatrogénica (ver secção 4.4).
Os pacientes devem ser informados sobre os sintomas dessas reações adversas potenciais e instruídos a informar seu médico se sentirem sinais como dor ocular ou aumento do desconforto, agravamento da vermelhidão ocular, visão turva ou diminuída, um número elevado de moscas volantes ou um " maior sensibilidade à luz.
As reações adversas notificadas após a administração de Lucentis em estudos clínicos estão resumidas na tabela abaixo.
Tabela de reações adversas #
As reações adversas são listadas por classe de sistemas de órgãos e frequência usando a seguinte convenção: muito comuns (≥1 / 10), comuns (≥1 / 100,
Infecções e infestações
Muito comum Nasofaringite
comum Infecção do trato urinário *
Doenças do sistema sanguíneo e linfático
comum Anemia
Distúrbios do sistema imunológico
comum Hipersensibilidade
Distúrbios psiquiátricos
comum Ansiedade
Doenças do sistema nervoso
Muito comum Dor de cabeça
Desordens oculares
Muito comum Vitreíte, descolamento de vítreo, hemorragia retiniana, distúrbios visuais, dor ocular, moscas volantes vítreas, hemorragia conjuntival, irritação ocular, sensação de corpo estranho no olho, lacrimação aumentada, blefarite, olho seco, hiperemia ocular, olho com coceira.
comum Degeneração da retina, distúrbios da retina, descolamento da retina, rasgo da retina, descolamento do epitélio pigmentar da retina, rasgo do epitélio pigmentar da retina, acuidade visual prejudicada, hemorragia vítrea, distúrbios vítreos, uveíte, irite, iridociclite, catarata, cápsula de catarata subcapsular punctátil posterior, ceratite, abrasão da córnea, reação da câmara anterior, visão turva, hemorragia no local da injeção, hemorragia ocular, conjuntivite, conjuntivite
alérgico, secreção ocular, flashes luminosos, fotofobia, desconforto ocular, edema palpebral, dor palpebral, hiperemia conjuntival.
Incomum Cegueira, endoftalmite, hipópio, hifema, ceratopatia, sinéquia da íris, depósitos na córnea, edema da córnea, estrias da córnea, dor no local da injeção, irritação no local da injeção, sensação anormal nos olhos, irritação da pálpebra.
Doenças respiratórias, torácicas e do mediastino
comum Tosse
Problemas gastrointestinais
comum Náusea
Afecções do tecido cutâneo e subcutâneo
comum Reações alérgicas (erupção na pele, urticária, coceira, eritema)
Afecções musculoesqueléticas e dos tecidos conjuntivos
Muito comum Artralgia
Testes de diagnóstico
Muito comum Pressão intraocular aumentada
# As reações adversas foram definidas como eventos adversos (em pelo menos 0,5 pontos percentuais de pacientes) que ocorreram em uma taxa mais elevada (pelo menos 2 pontos percentuais) em pacientes recebendo tratamento com Lucentis 0,5 mg em comparação com aqueles que receberam tratamento de controle (sham ou PDT verteporfina).
* observado apenas na população com DME
Reações adversas relacionadas à categoria de medicamentos
Nos estudos de fase III de DMRI úmida, a frequência geral de hemorragias não oculares, um evento adverso potencialmente relacionado aos inibidores de VEGF (fator de crescimento dos vasos endoteliais), foi ligeiramente aumentada em pacientes tratados com ranibizumabe. No entanto, não foi. padrão entre as diferentes hemorragias. Existe um risco teórico de eventos tromboembólicos arteriais, incluindo acidente vascular cerebral e infarto do miocárdio, resultante do uso intravítreo de inibidores de VEGF.Uma baixa incidência de eventos tromboembólicos arteriais foi observada em ensaios clínicos com Lucentis em pacientes com DMRI, DME, RVO e PM e nenhuma diferença foi observada entre os grupos de ranibizumabe em comparação com o controle.
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas que ocorram após a autorização do medicamento é importante, uma vez que permite a monitorização contínua da relação benefício / risco do medicamento.Os profissionais de saúde são convidados a notificar quaisquer suspeitas de reações adversas através da Agência Italiana de Medicamentos. , site: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Overdose
Foram notificados casos de sobredosagem acidental em ensaios clínicos em DMRI húmida e dados pós-comercialização. As reações adversas mais frequentemente associadas a estes casos foram aumento da pressão intraocular, cegueira transitória, diminuição da acuidade visual, edema da córnea e dor. Se ocorrer uma sobredosagem, a pressão intraocular deve ser monitorada e tratada conforme necessário pelo médico.
05.0 PROPRIEDADES FARMACOLÓGICAS
05.1 Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Oftalmológicos, agentes antineovasculares, código ATC: S01LA04
Ranibizumab é um fragmento de anticorpo monoclonal recombinante humanizado dirigido contra o fator de crescimento endotelial vascular humano A (VEGF-A). Liga-se com alta afinidade às isoformas de VEGF-A (por exemplo, VEGF110, VEGF121 e VEGF165), evitando assim a ligação de VEGF-A aos seus receptores VEGFR-1 e VEGFR-2. uma neovascularização e um aumento na permeabilidade vascular, que se acredita que contribuem para a progressão da forma neovascular de degeneração macular relacionada à idade, miopia patológica ou diminuição da visão causada por edema macular diabético ou "Edema macular secundário a RVO.
Tratamento da DMRI úmida
Para a DMRI úmida, a segurança e a eficácia clínica de Lucentis foram avaliadas em três estudos randomizados de 24 meses, duplo-cegos, com controle simulado ou ativo em pacientes com DMRI neovascular. Um total de 1.323 pacientes (879 tratados e 444 controles) foram incluídos nesses estudos.
No estudo FVF2598g (MARINA), 716 pacientes com lesões de neovascularização coroidal (CNV) minimamente clássica ou oculta sem componente clássico receberam injeções intravítreas mensais de Lucentis 0,3 mg (n = 238) ou 0,5 mg (n = 240) ou injeções simuladas (n = 238).
No estudo FVF2587g (ANCHOR), 423 pacientes com CNV predominantemente clássico receberam um dos seguintes tratamentos: 1) injeções intravítreas mensais de Lucentis 0,3 mg e simulação de PDT (n = 140); 2) injeções intravítreas mensais de Lucentis 0,5 mg e simulação de PDT (n = 140); ou 3) injeções sham intravítreas e PDT com verteporfina (n = 143). PDT com verteporfina ou sham foi administrado junto com a injeção inicial de Lucentis e, posteriormente, a cada 3 meses se a fluorangiografia mostrasse persistência ou retomada do vazamento vascular.
As principais conclusões estão resumidas nas Tabelas 1, 2 e Figura 1.
Tabela 1 Resultados no mês 12 e mês 24 no estudo FVF2598g (MARINA)
ap
Tabela 2 Resultados no Mês 12 e Mês 24 no Estudo FVF2587g (ANCHOR)
Mesa
Os resultados de ambos os estudos mostraram que o tratamento contínuo com ranibizumabe também pode ser benéfico em pacientes que perderam ≥15 letras da melhor acuidade visual corrigida (BCVA) no primeiro ano de tratamento.
O estudo FVF3192g (PIER) foi um estudo randomizado, duplo-cego e controlado por sham, projetado para avaliar a segurança e eficácia de Lucentis em 184 pacientes com todas as formas de DMRI neovascular. Os pacientes receberam injeções intravítreas de Lucentis 0,3 mg (n = 60) ou 0,5 mg (n = 61) ou injeções sham (n = 63) uma vez por mês por 3 doses consecutivas, seguidas por uma dose administrada uma vez a cada 3 meses. A partir do mês 14 do estudo, os pacientes tratados com uma injeção sham foram admitidos em tratamento com ranibizumab e a partir do mês 19, tratamentos mais frequentes podem ser realizados. Os pacientes tratados com Lucentis no estudo PIER receberam uma média de 10 tratamentos no total.
O endpoint primário de eficácia foi a alteração média na acuidade visual em 12 meses em comparação com a linha de base. Após um aumento inicial na acuidade visual (após a dose mensal), em média, a acuidade visual dos pacientes diminuiu com a dosagem trimestral, retornando à linha de base no mês 12 e este efeito foi mantido na maioria dos pacientes tratados. Com ranibizumabe (82%) em Mês 24. Os dados de um número limitado de indivíduos que foram transferidos para o tratamento com ranibizumabe após mais de um ano de tratamento simulado sugeriram que o início precoce do tratamento pode estar associado a uma melhor retenção da "acuidade visual.
Em ambos os estudos MARINA e ANCHOR, a melhora na acuidade visual observada com Lucentis 0,5 mg em 12 meses foi acompanhada por benefícios relatados pelo paciente, conforme medido pela pontuação do National Eye Institute Visual Function Questionnaire (VFQ-25). Diferenças entre Lucentis 0,5 mg e os dois grupos de controle foram avaliados com valores de p variando de 0,009 a
A eficácia de Lucentis no tratamento de DMRI úmida foi confirmada em estudos de DMRI pós-comercialização. Dados de dois estudos (MONT BLANC, BPD952A2308 e DENALI, BPD952A2309) não demonstraram efeitos adicionais. Da administração combinada de verteporfina (Visudyne PDT) e Lucentis comparados para Lucentis sozinho.
Tratamento da deficiência visual devido a DME
A segurança e eficácia de Lucentis foram avaliadas em dois estudos randomizados, duplo-cegos, controlados por sham ou ativos de 12 meses em pacientes com visão diminuída devido a edema macular diabético. Um total desses estudos foi inscrito. De 496 pacientes (336 ativos e 160 controles), a maioria tinha diabetes tipo II, 28 pacientes tratados tinham diabetes tipo I.
Na fase II do estudo D2201 (RESOLVE), 151 pacientes foram tratados com ranibizumabe (6 mg / ml, n = 51, 10 mg / ml, n = 51) ou simulação (n = 49) com uma "injeção intravítrea por mês. até que os critérios predefinidos fossem atendidos. A dose inicial de ranibizumabe (0,3 mg ou 0,5 mg) pode ser dobrada a qualquer momento durante o estudo após a primeira injeção. A fotocoagulação a laser foi permitida como tratamento de resgate a partir do mês 3 em ambos os braços de tratamento. teve duas partes: uma parte exploratória (os primeiros 42 pacientes visitados no mês 6) e uma parte confirmatória (os 109 pacientes restantes visitados no mês 12).
Os principais achados da parte confirmatória do estudo (2/3 dos pacientes) estão resumidos na Tabela 3.
Tabela 3 Resultados no 12º mês no Estudo D2201 (RESOLVE) (População total do estudo)
ap
No estudo de fase III D2301 (RESTORE), 345 pacientes com deficiência visual devido a edema macular foram randomizados para receber uma "injeção intravítrea de 0,5 mg de ranibizumabe como monoterapia e fotocoagulação simulada a laser (n = 116) ou uma combinação de 0,5 mg de ranibizumabe e fotocoagulação a laser (n = 118) ou uma injeção simulada e fotocoagulação a laser (n = 111). O tratamento com ranibizumabe foi iniciado com injeções intravítreas mensais e continuou até que a acuidade visual permanecesse estável por pelo menos três verificações mensais consecutivas. O tratamento foi retomado quando foi observada uma redução no BCVA devido à progressão de DME. Fotocoagulação a laser foi administrada no início do mesmo dia, pelo menos 30 minutos antes da injeção de ranibizumabe e, posteriormente, conforme necessário com base nos critérios ETDRS.
As principais conclusões estão resumidas na Tabela 4 e na Figura 2.
Tabela 4 Resultados no mês 12 no Estudo D2301 (RESTORE)
ap
O efeito foi consistente na maioria dos subgrupos. No entanto, indivíduos com um BCVA bastante alto na linha de base (> 73 letras) com edema macular e espessura central da retina
A melhora na acuidade visual no 12º mês observada com Lucentis 0,5 mg foi acompanhada por benefícios relatados pelo paciente das principais funções relacionadas à visão, conforme medido pela pontuação do National Eye Institute Visual Function Questionnaire (VFQ-25). Nenhuma diferença devido ao tratamento poderia ser estabelecido nas subclasses deste questionário. A diferença entre Lucentis 0,5 mg e o grupo controle foi avaliada com um valor de p de 0,0137 (ranibizumabe mono) e 0,0041 (ranibizumabe + laser) para o escore composto do VFQ-25.
Em ambos os estudos, a melhora visual foi acompanhada por uma redução contínua do edema macular medido como espessura retinal central (CRT).
Tratamento da deficiência visual causada por edema macular secundário a RVO
A segurança clínica e a eficácia de Lucentis em pacientes com deficiência visual devido a edema macular secundário a RVO foram avaliadas em ensaios clínicos randomizados, duplo-cegos controlados: BRAVO e CRUISE que recrutaram pacientes com BRVO (n = 397) e CRVO (n = 392 ). Em ambos os estudos, os pacientes receberam 0,3 mg ou 0,5 mg de ranibizumabe intravítreo ou injeções sham. Após 6 meses, os pacientes no braço de controle sham foram transferidos para o grupo de ranibizumabe 0,5 mg. No estudo BRAVO, fotocoagulação a laser como tratamento de resgate foi permitido em todos os braços a partir do mês 3.
As principais conclusões dos estudos BRAVO e CRUISE são apresentadas nas Tabelas 5 e 6
Tabela 5 Resultados no mês 6 e 12 (BRAVO)
ap
Tabela 6 Resultados no mês 6 e 12 (CRUZEIRO)
ap
Em ambos os estudos, a melhora visual foi acompanhada por uma redução contínua e significativa do edema macular medido em termos de espessura retiniana central.
Em pacientes BRVO (estudo BRAVO e extensão do estudo HORIZON): Após 2 anos, os pacientes que foram tratados com injeções simuladas nos primeiros 6 meses e posteriormente mudaram para o tratamento com ranibizumabe tiveram um ganho de AV (& symp; 15 letras) comparável àquele de pacientes que haviam sido tratados com ranibizumabe desde o início do estudo (& symp; 16 letras). No entanto, o número de pacientes que completaram 2 anos foi limitado e apenas visitas trimestrais foram agendadas no estudo HORIZON. há evidências suficientes para concluir com recomendações sobre quando o tratamento com ranibizumabe deve ser iniciado em pacientes com BRVO.
Em pacientes CRVO (estudo CRUISE e extensão do estudo HORIZON): Após 2 anos, os pacientes que foram tratados nos primeiros 6 meses com injeções simuladas e subsequentemente transferidos para tratamento com ranibizumabe não mostraram ganhos em AV (& symp; 6 letras) em comparação com aqueles de pacientes que haviam sido tratados com ranibizumabe desde o início do estudo (& symp; 12 letras).
A melhora na acuidade visual observada com o tratamento com ranibizumabe nos meses 6 e 12 foi acompanhada por benefícios relatados pelo paciente, conforme medido pelo subgrupo do Questionário de Função Visual do National Eye Institute (NEI VFQ-25) de atividades próximas e distantes. A diferença entre Lucentis 0,5 mg e o grupo controle ficou entre os valores de p entre 0,02 e 0,0002.
Tratamento da deficiência visual devido a CNV secundária a PM
A segurança e eficácia clínica de Lucentis em pacientes com deficiência visual devido a CNV em PM foram validadas com base em dados de 12 meses do estudo principal randomizado, duplo-cego e controlado F2301 (RADIANCE). Este estudo teve como objetivo avaliar dois regimes de dosagem diferentes de ranibizumabe 0,5 mg administrado por injeção intravítrea versus PDT com verteporfina (vPDT, terapia fotodinâmica com Visudyne). Os 277 pacientes foram randomizados para um dos seguintes braços:
• Grupo I (ranibizumabe 0,5 mg, regime de tratamento determinado por critérios de "estabilidade" definidos como nenhuma alteração na BCVA em comparação com as avaliações dos dois meses anteriores).
• Grupo II (ranibizumabe 0,5 mg, regime de tratamento determinado pelos critérios de "atividade da doença" definidos como deficiência visual atribuível ao fluido intra ou sub-retiniano ou vazamento ativo causado por lesões CNV conforme evidenciado por OCT e / ou AF).
• Grupo III (pacientes tratados com vPDT - com possibilidade de tratamento com ranibizumab a partir do mês 3).
Durante os 12 meses do estudo, os pacientes receberam uma média de 4,6 injeções (variação de 1-11) no Grupo I e 3,5 injeções (variação de 1-12) no Grupo II. Entre os pacientes pertencentes ao Grupo II, que reflete a posologia recomendada (ver seção 4.2), 50,9% dos pacientes foram submetidos a tratamento com 1 a 2 injeções, 34,5% 3 a 5 injeções e 14,7% deram 6 a 12 injeções durante o estudo de 12 meses . 62,9% dos pacientes do Grupo II não necessitaram de injeções durante os segundos 6 meses do estudo.
Os principais resultados do RADIANCE estão resumidos na Tabela 7 e na Figura 5.
Tabela 7 Resultados no mês 3 e 12 (RADIANCE)
ap
b Controle comparativo até o mês 3. Os pacientes randomizados para receber vPDT eram elegíveis para o tratamento com ranibizumabe no mês 3 (no Grupo III, 38 pacientes receberam ranibizumabe no mês 3)
A melhora na visão foi acompanhada por uma redução na espessura central da retina.
Em comparação com o grupo tratado com vPDT, os pacientes nos grupos tratados com ranibizumabe relataram benefício (valor p
População pediátrica
A segurança e eficácia de ranibizumab em crianças ainda não foram estabelecidas.
A Agência Europeia de Medicamentos dispensou a obrigação de apresentação dos resultados dos estudos com Lucentis em todos os subconjuntos da população pediátrica para DMRI neovascular, deficiência visual devido a DME, deficiência visual devido a edema macular secundário a RVO e deficiência visual devido a NVC secundária a PM (ver seção 4.2 para informações sobre uso pediátrico).
05.2 Propriedades farmacocinéticas
Após a administração intravítrea mensal de Lucentis a pacientes com DMRI neovascular, as concentrações séricas de ranibizumabe foram geralmente baixas, com níveis de pico (Cmax) geralmente abaixo da concentração de ranibizumabe necessária para inibir a atividade biológica de VEGF em 50% (11-27 ng / mL, avaliado em um teste em vitro proliferação celular). Cmax foi proporcional à dose em todo o intervalo de dose de 0,05 a 1,0 mg / olho. Em um número limitado de pacientes com DME, as concentrações séricas detectadas indicam que uma exposição sistêmica ligeiramente maior não pode ser excluída do que aquelas observadas em pacientes com DMRI neovascular. As concentrações séricas de ranibizumabe em pacientes com RVO foram semelhantes ou ligeiramente superiores às observadas em pacientes com DMRI neovascular.
Com base na análise farmacocinética populacional e na depuração sérica de ranibizumabe para pacientes com DMRI neovascular tratados com a dose de 0,5 mg, a meia-vida de eliminação vítrea média de ranibizumabe é de aproximadamente 9 dias. No momento da administração intravítrea mensal de Lucentis 0,5 mg / olho, o C sérico de ranibizumabe, atingiu aproximadamente 1 dia após a dose, geralmente varia entre 0,79 e 2,90 ng / ml, enquanto se espera que a Cmin geralmente flutue entre 0,07 e 0,49 ng / ml.As concentrações séricas de ranibizumabe são estimadas em aproximadamente 90.000 vezes mais baixas do que as concentrações no vítreo.
Doentes com insuficiência renal: Não foram realizados estudos convencionais para examinar a farmacocinética de Lucentis em doentes com insuficiência renal. Em uma "análise farmacocinética em uma população de pacientes com DMRI neovascular, 68% (136 em 200) dos pacientes tinham" insuficiência renal (46,5% leve [50-80 mL / min], 20% moderada [30 -50 mL / min] e 15% grave [a depuração sistêmica foi ligeiramente menor, mas isso não foi clinicamente significativo.
Doentes com insuficiência hepática: Não foram realizados estudos convencionais para examinar a farmacocinética de Lucentis em doentes com insuficiência hepática.
05.3 Dados de segurança pré-clínica
A administração intravítrea bilateral de ranibizumabe a macacos cynomolgus em doses entre 0,25 mg / olho e 2,0 mg / olho uma vez a cada 2 semanas por até 26 semanas resultou em efeitos oculares dependentes da dose.
Intraocularmente, aumentos dependentes da dose em exacerbação e células ocorreram na câmara anterior, com pico 2 dias após a injeção. A gravidade da resposta inflamatória geralmente diminui com injeções subsequentes ou durante o período de recuperação. No segmento posterior ocorreram infiltrações celulares e moscas volantes. que também tendeu a ser dependente da dose e geralmente persistiu até o final do período de tratamento. No estudo de 26 semanas, a gravidade da inflamação vítrea aumentou com o número de injeções. No entanto, a reversibilidade foi observada após o período de recuperação. A natureza e a duração da inflamação do segmento posterior são indicativas de uma resposta imunomediada por anticorpos, que pode ser clinicamente irrelevante. A formação de catarata foi observada em alguns animais após um período relativamente longo de inflamação intensa, sugerindo que as alterações do cristalino eram secundárias a inflamação grave. Um aumento transitório na pressão intraocular foi observado após a administração, independentemente da dose, após injeções intravítreas.
Alterações microscópicas oculares estavam relacionadas à inflamação e não indicavam processos degenerativos. Alterações granulomatosas inflamatórias foram observadas no disco óptico de alguns olhos. Essas alterações do segmento posterior diminuíram e, em alguns casos, desapareceram, durante o período de recuperação.
Não houve sinais de toxicidade sistêmica após a administração intravítrea. Anticorpos séricos e vítreos para ranibizumabe foram encontrados em um subconjunto de animais tratados.
Não há dados de carcinogenicidade ou mutagenicidade disponíveis.
Em macacas grávidas, a injeção intravítrea de ranibizumabe, resultando em uma exposição sistêmica máxima 0,9-7 vezes a pior exposição clínica, não causou toxicidade no desenvolvimento ou teratogenicidade, e não teve efeito sobre o peso ou a estrutura do corpo. Placenta, embora o ranibizumabe deva ser considerado potencialmente teratogênica e embrio / fetotóxica com base em seu efeito farmacológico.
A ausência de efeitos mediados do ranibizumab no desenvolvimento embrionário / fetal está plausivelmente relacionada principalmente à incapacidade do fragmento Fab de atravessar a placenta. No entanto, foi descrito um caso com níveis séricos maternos elevados de ranibizumabe e a presença de ranibizumabe no soro fetal, sugerindo que o anticorpo anti-ranibizumabe atuou como uma proteína (contendo a região FC) que transporta ranibizumabe, diminuindo assim sua eliminação do soro materno e permitindo sua transferência para a placenta. Uma vez que os testes de desenvolvimento embrio / fetal foram conduzidos em animais saudáveis grávidas e algumas doenças (como diabetes) podem modificar a permeabilidade da placenta a um fragmento Fab, o estudo deve ser interpretado com cautela.
06.0 INFORMAÇÕES FARMACÊUTICAS
06.1 Excipientes
α, α-trealose di-hidratada
Cloridrato de histidina, monohidrato
Histidina
Polissorbato 20
Água para injetáveis
06.2 Incompatibilidade
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros medicamentos.
06.3 Período de validade
3 anos
06.4 Precauções especiais de armazenamento
Conservar no frigorífico (2 ° C - 8 ° C).
Não congele.
Manter o frasco para injectáveis dentro da embalagem exterior para proteger o medicamento da luz.
06.5 Natureza da embalagem primária e conteúdo da embalagem
0,23 ml de solução estéril em um frasco (vidro tipo I) com uma rolha (borracha de clorobutila), 1 agulha de filtro romba (18G x 1½ ", 1,2 mm x 40 mm, 5 mcm), 1 agulha de injeção (30G x ½", 0,3 mm x 13 mm) e 1 seringa (polipropileno) (1 ml). A embalagem contém 1 frasco para injetáveis.
06.6 Instruções de uso e manuseio
O frasco para injetáveis, a agulha para injeção, a agulha com filtro e a seringa destinam-se a uma única utilização. A reutilização pode causar infecção ou outras doenças / lesões. Todos os componentes são estéreis. Qualquer componente com embalagem mostrando sinais de danos ou adulteração não deve ser usado. A esterilidade não pode ser garantida se o selo da embalagem do componente não estiver intacto.
Para preparar Lucentis para injeção intravítrea, siga as instruções abaixo:
1. Desinfete a parte externa da tampa de borracha do frasco antes da coleta.
2. Conecte assepticamente a agulha com filtro de 5 mcm (18G x 1½ ", 1,2 mm x 40 mm, fornecida) a uma seringa de 1 ml (fornecida). Insira a agulha de filtro romba no centro da tampa até tocar o fundo do frasco.
3. Retire todo o líquido do frasco, segurando-o na posição vertical, ligeiramente inclinado para facilitar a retirada completa.
4. Certifique-se de que o êmbolo da seringa está puxado para trás o suficiente ao esvaziar o frasco para esvaziar completamente a agulha com filtro.
5. Deixe a agulha com filtro embotada no frasco para injetáveis e retire a seringa de dentro.Deite fora a agulha com filtro após retirar o conteúdo do frasco para injetáveis e não a utilize para injeção intravítrea.
6. Fixe a agulha de injeção (30G x ½ ", 0,3 mm x 13 mm, fornecida) de forma segura e asséptica à seringa.
7. Remova cuidadosamente a tampa da agulha de injeção sem desconectar a agulha de injeção da seringa.
Nota: Segure a base amarela da agulha de injeção enquanto remove a tampa.
8. Expulse cuidadosamente o ar da seringa e ajuste a dose para 0,05 ml marcado na seringa.A seringa está pronta para a injeção.
Nota: Não limpe a agulha de injeção.Não puxe o êmbolo.
Após a injeção, não cubra a agulha nem a destaque da seringa. Descarte a seringa usada junto com a agulha em um recipiente apropriado ou de acordo com as exigências locais.
07.0 TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
Reino Unido
08.0 NÚMERO DE AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
EU / 1/06/374/001
037608027
09.0 DATA DA PRIMEIRA AUTORIZAÇÃO OU RENOVAÇÃO DA AUTORIZAÇÃO
Data da primeira autorização: 22 de janeiro de 2007
Data da renovação mais recente: 24 de janeiro de 2012
10.0 DATA DE REVISÃO DO TEXTO
05/2014













.jpg)